Острый лейкоз — Википедия
Острый лейкоз — клональное (онкологическое) заболевание, первично возникающее в костном мозге в результате мутации стволовой клетки крови. Следствием мутации является потеря потомками мутировавшей клетки способности к дифференцировке до зрелых клеток крови. Морфологический субстрат острыx лейкозов — бластные клетки.
Как и для большинства других опухолевых заболеваний, для острых лейкозов невозможно выделить специфический этиологический фактор.
Острые лейкозы делятся на ряд видов, среди которых наибольшее значение имеют острый лимфобластный и острый миелобластный лейкозы.
Необходимо отметить, что острый лейкоз никогда не переходит в хронический, а хронический никогда не обостряется — таким образом, термины «острый» и «хронический» используются только из-за удобства; значение этих терминов в гематологии отличается от значения в других медицинских дисциплинах. Исключением является лишь хронический миелобластный лейкоз, острая или терминальная фаза которого характеризуется развитием бластного криза — появлением в крови и костном мозге 30-90 % бластных клеток, то есть развитием острого миелобластного (или лимфобластного) лейкоза.
- Острые миелобластные лейкозы (ОМЛ)
- Острый малодифференцированный лейкоз
- ОМЛ без созревания
- ОМЛ с созреванием
- Острый промиелобластный лейкоз
- Острый миеломонобластный лейкоз
- Острый монобластный лейкоз
- Острый эритромиелоз
- Острый мегакариобластный лейкоз
- Острые лимфобластные лейкозы (ОЛЛ)
- Пре-пре-B-ОЛЛ
- Пре-B-ОЛЛ
- B-ОЛЛ
- T-ОЛЛ
Ежегодно регистрируется 35 новых случаев острых лейкозов на 1 млн населения. Структура заболеваемости в значительной степени зависит от возраста. ОЛЛ чаще развивается в детском возрасте и после 40 лет. Частота ОМЛ одинакова во всех возрастных rpуппах. Мужчины и женщины болеют с одинаковой частотой.
В основе патогенеза острых лейкозов лежит мутация стволовой клетки крови, что влечёт за собой практически полную потерю потомками мутировавшей клетки способности к созреванию. Мутантный клон автономен от каких-либо регулирующих воздействий организма и довольно быстро вытесняет нормальные гемопоэтические клетки, замещая собой весь гемопоэз.Степень злокачественности опухолевых клеток при острых лейкозах с течением времени возрастает (как и для других групп опухолей, для острых лейкозов правомочен закон опухолевой прогрессии). Поскольку опухолевые клетки при острых лейкозах в большинстве вариантов изначально имеют выраженный дефект созревания, то большая злокачественность часто проявляется возникновением экстрамедуллярных очагов кроветворения, увеличением пролиферативной активности, развитием резистентности к проводимой терапии.
Следствием мутации стволовой клетки является развитие в костном мозге клона клеток, утративших способность к созреванию. Неопластический клон вытесняет нормальные гемопоэтические клетки, что приводит к развитию дефицита зрелых клеток в периферической крови. Снижение количества или полное отсутствие зрелых клеток периферической крови обусловливает выпадение соответствующих функций периферической крови, что влечёт за собой развитие клинических симптомов заболевания.
Различные формы острого лейкоза имеют стереотипные морфологические проявления: лейкозная инфильтрация костного мозга в виде очаговых и диффузных инфильтратов из клеток с крупными светлыми ядрами, содержащими по нескольку ядрышек. Размеры и очертания ядер, а также ширина ободка плазмы могут варьировать. Бласты составляют 10-20 % мозговых клеток. Цитогенетическую принадлежность бластов, как правило, можно выявить только с помощью специальных методов исследования — цитохимических и иммуногистохимических. Применяются реакции на пероксидазу, окраска на липиды суданом чёрным, ШИК-реакция, гистоферментохимические акции на выявление неспецифической эстеразы, хлорацетатэстеразы, кислой фосфатазы. Иммуногистохимически возможно определение маркеров B-, T-лимфоцитов, клеток миелоидного и моноцитарного рядов..
В периферической крови и в костном мозге описывается феномен «лейкемического провала» («hiatus leucemicus»), развивающийся за счёт наличия только бластных и дифференцированных клеток и отсутствия промежуточных форм.
В костномозговой ткани происходят вытеснение нормальных клеток гемопоэза опухолевыми, истончение и резорбция ретикулярных волокон, нередко развивается миелофиброз. При цитостатической терапии происходит опустошение костного мозга с гибелью бластных форм, увеличивается число жировых клеток и разрастается соединительная ткань.
Лейкозные инфильтраты в виде диффузных или очаговых скоплений обнаруживаются в лимфатических узлах, селезёнке и печени. Это приводит к увеличению размеров этих органов. В печени характерно развитие жировой дистрофии. Возможна лейкозная инфильтрация слизистых оболочек полости рта и ткани миндалин.
Клинические проявления одинаковы для всех вариантов острых лейкозов и могут быть довольно полиморфными. Начало заболевания может быть внезапным или постепенным. Для них не существует характерного начала, каких-либо специфических клинических признаков. Только тщательный анализ клинической картины позволяет распознать скрывающееся под видом «банального» заболевания более серьёзное.
Характерна комбинация синдромов недостаточности костного мозга и признаков специфического поражения.
В связи с лейкозной инфильтрацией слизистых оболочек полости рта и ткани миндалин появляются некротический гингивит, тонзиллит (некротическая ангина). Иногда присоединяется вторичная инфекция и развивается сепсис, приводящий к смерти.
Тяжесть состояния больного может быть обусловлена выраженной интоксикацией, геморрагическим синдромом, дыхательной недостаточностью (вследствие сдавления дыхательных путей увеличенными внутригрудными лимфатическими узлами).
Использование активной цитостатической терапии повлияло на течение острых лейкозов, то есть привело к индуцированному лекарственному патоморфозу. В связи с этим в настоящее время выделяют следующие клинические стадии заболевания:
- первая атака,
- ремиссия (полная или неполная),
- рецидив (первый, повторный).
Недостаточность костного мозга[править | править код]
Она проявляется в виде инфекционных осложнений, ДВС-синдрома, геморрагического и анемического синдромов.
Развитие инфекционных осложнений происходит вследствие иммунодефицита, вызванного нарушением функции лейкоцитов. Чаще всего инфекционные осложнения имеют бактериальное происхождение, грибковые и вирусные инфекции встречаются реже. Могут развиться ангина, гингивит, стоматит, остеомиелиты челюстно-лицевой области, пневмония, бронхит, абсцессы, флегмоны, сепсис.
Геморрагический синдром при острых лейкозах обусловлен тромбоцитопенией, повреждением печени и стенок сосудов. Он проявляется геморрагическим диатезом петехиально-пятнистого типа. На коже и слизистых оболочках появляются «синячки» и петехии небольшого размера. Появление геморрагий легко провоцируется самыми незначительными воздействиями — трением одежды, легкими ушибами. Могут иметь место носовые кровотечения, кровотечения из десен, метроррагии, кровотечения из мочевыводящих путей. Геморрагический синдром может привести к весьма опасным осложнениям — кровоизлияниям в головной мозг и желудочно-кишечным кровотечениям.
Анемический синдром проявляется в виде бледности, одышки, учащённое сердцебиения, сонливости.
ДВС-синдром чаще имеет место при промиелоцитарном лейкозе.
Специфическое поражение[править | править код]
Отмечаются признаки интоксикации: снижение массы тела, лихорадка, слабость, потливость, снижение аппетита.
Может наблюдаться инфильтрация десен лейкозными клетками, при этом десны гиперплазированы, нависают над зубами, гиперемированы.
Пролиферативный синдром может проявляться увеличением размеров лимфатических узлов (лимфоаденопатия), селезёнки, печени. В ряде случаев на коже появляются лейкемиды — приподнимающиеся над поверхностью кожи образования мягкой или плотной консистенции. Цвет их может соответствовать цвету кожи или быть светло-коричневым, жёлтым, розовым.
Поражение ЦНС (нейролейкемия) возникает особенно часто при ОЛЛ и значительно ухудшает прогноз. Возникает метастазирование лейкозных клеток в оболочки головного и спинного мозга или в вещество мозга. Клинически возможны проявления различной тяжести — от головной боли до тяжелых очаговых поражений.
Манифестация острого лейкоза может быть внезапной или стертой.
- Патологическая анатомия. Курс лекций. Под ред. В. В. Серова, М. А. Пальцева. — М.: Медицина, 1998
- Шулутко Б. И., Макаренко С. В. Стандарты диагностики и лечения внутренних болезней. 3-е изд. СПб.: «Элби-СПБ», 2005
Фонд больных лейкемией. Литература. Статьи. http://www.leukaemia.org.au/
Европейский банк доноров стволовых клеток. http://www.stefan-morsch-stiftung.com/
Описание лейкозов в Большой Медицинской Энциклопедии. http://www.neuronet.ru/bibliot/bme/des/des176.html
Гематологический Научный центр РАМН http://www.blood.ru/
Сайт поддержки больных лейкозом https://web.archive.org/web/20140715001917/http://onelife.guchua.com/
Хронический миелоидный лейкоз — Википедия
Хронический миелоидный лейкоз (ХМЛ, хронический миелолейкоз) — форма лейкоза, которая характеризуется ускоренной и нерегулируемой пролиферацией преимущественно миелоидных клеток в костном мозге с их накоплением в крови. ХМЛ — гемопоэтическое клональное заболевание, основным проявлением которого является пролиферация зрелых гранулоцитов (нейтрофилов, эозинофилов и базофилов) и их предшественников. Это миелопролиферативное заболевание ассоциировано с характерной хромосомной транслокацией (филадельфийской хромосомой). В настоящее время основным способом лечения хронического миелолейкоза является таргетная (целевая) терапия ингибиторами тирозинкиназ, такими как иматиниб, нилотиниб, дазатиниб и другие, значительно улучшившая показатели выживаемости.
На примере ХМЛ впервые была показана связь злокачественного заболевания с конкретной генетической аномалией. В случае ХМЛ такой характерной аномалией является хромосомная транслокация, которая проявляется присутствием в кариотипе так называемой филадельфийской хромосомы. Эта мутантная хромосома получила своё название по месту работы её первооткрывателей, Питера Ноуелла (Пенсильванский университет) и Дэвида Хангерфорда (Онкологический центр Фокса Чейза), которые впервые описали её в 1960 году в Филадельфии (штат Пенсильвания, США)[2].
При этой транслокации, участки 9-й и 22-й хромосом меняются местами. В результате, фрагмент гена BCR из хромосомы 22 и ген ABL из хромосомы 9 образуют единую рамку считывания. Продуктами этого аномального слитого гена могут быть белки с молекулярной массой 210 (p210) или, реже, 185 кДа (p185). Так как в норме белок ABL содержит тирозинкиназный домен, продукт мутантного гена также является тирозинкиназой
Белок BCR-ABL взаимодействует с одной из субъединиц клеточного рецептора к интерлейкину 3. Транскрипция гена BCR-ABL происходит непрерывно и не нуждается в активации другими белками. BCR-ABL активирует сигнальный каскад, контролирующий клеточный цикл, ускоряя деление клеток. Более того, белок BCR-ABL подавляет репарацию ДНК, вызывая неустойчивость генома и делая клетку более восприимчивой к дальнейшим генетическим аномалиям. Активность BCR-ABL — патофизиологическая причина хронического миелолейкоза. По мере изучения природы белка BCR-ABL и его тирозинкиназной активности, была разработана таргетная (целевая) терапия, позволяющая специфически ингибировать эту активность. Ингибиторы тирозинкиназ могут способствовать полной ремиссии ХМЛ, что ещё раз подтверждает ведущую роль BCR-ABL в развитии заболевания[4].
Заболевание часто протекает бессимптомно, выявляясь при рутинном клиническом анализе крови. В этом случае ХМЛ следует дифференцировать от лейкемоидной реакции, при которой мазок крови может иметь схожую картину. ХМЛ может проявляться недомоганием, субфебрильной лихорадкой, подагрой, повышенной восприимчивостью к инфекциям, анемией, тромбоцитопенией с кровоточивостью (хотя также может наблюдаться повышенное содержание тромбоцитов). Также отмечается спленомегалия.[3][5]
В течение ХМЛ выделяют три фазы на основании клинических характеристик и лабораторных данных. В отсутствие лечения ХМЛ обычно начинается с хронической фазы, в течение нескольких лет прогрессирует в фазу акселерации и, в конечном счёте, завершается бластным кризом. Бластный криз — терминальная фаза ХМЛ, клинически подобная острому лейкозу. Вовремя начатое медикаментозное лечение, как правило, может остановить прогрессирование болезни по этому пути. Одним из факторов прогрессии от хронической фазы к бластному кризу является приобретение новых хромосомных аномалий (в дополнение к филадельфийской хромосоме)[3]. Некоторые пациенты к моменту постановки диагноза могут находиться уже в фазе акселерации или бластного криза[5].
Хроническая фаза[править | править код]
Около 85 % пациентов с ХМЛ к моменту постановки диагноза находятся в хронической фазе. В течение этой фазы клинические проявления обычно отсутствуют или имеются «лёгкие» симптомы, такие как недомогание или чувство переполнения живота. Продолжительность хронической фазы различна и зависит от того, насколько рано было диагностировано заболевание, а также от проведённого лечения. В конечном счёте, при отсутствии эффективного лечения, заболевание переходит в фазу акселерации[5].
Фаза акселерации[править | править код]
Диагностические критерии перехода в фазу акселерации могут различаться: наиболее широко используются критерии, установленные исследователями онкологического центра Андерсона при Техасском университете[6], Сокалом с соавторами[7], а также Всемирной организацией здравоохранения[8][9]. Критерии ВОЗ, вероятно, наиболее широко распространены, и отличают фазу акселлерации по следующим признакам:
- 10—19 % миелобластов в крови или костном мозге;
- > 20 % базофилов в крови или костном мозге;
- < 100 000 тромбоцитов в микролитре крови, вне связи с терапией;
- > 1 000 000 тромбоцитов в микролитре крови, вне зависимости от терапии;
- цитогенетическая эволюция с развитием новых аномалий в дополнение к филадельфийской хромосоме;
- прогрессирование спленомегалии или увеличение числа лейкоцитов, вне зависимости от терапии.
Фаза акселерации предполагается при наличии любого из указанных критериев. Фаза акселерации указывает на прогрессию заболевания и приближение бластного криза[8]
Бластный криз[править | править код]
Бластный криз — финальная стадия развития ХМЛ, протекающая, подобно острому лейкозу, с быстрой прогрессией и непродолжительной выживаемостью[5]. Бластный криз диагностируется на основе одного из следующих признаков у пациента с ХМЛ[10]:
- >20 % миелобластов или лимфобластов в крови или костном мозге;
- крупные группы бластов в костном мозге при биопсии;
- развитие хлоромы (солидного фокуса лейкемии вне костного мозга).
Предположение о ХМЛ часто делается на основании общего анализа крови, демонстрирующего повышение количества гранулоцитов всех типов, включая зрелые миелоидные клетки. Количество базофилов и эозинофилов повышено практически всегда, что позволяет дифференцировать ХМЛ и лейкемоидную реакцию. При диагностике ХМЛ часто проводится биопсия костного мозга, однако одной лишь морфологической оценки костного мозга недостаточно для постановки диагноза ХМЛ[4][5].
В конечном счёте, ХМЛ диагностируется посредством выявления филадельфийской хромосомы в образцах костного мозга. Эта характерная хромосомная аномалия может быть выявлена в результате цитогенетического анализа, при помощи флюоресцентной гибридизации in situ или детекции гена BCR-ABL методом ПЦР[5].
Существуют разногласия в отношении так называемого Ph-негативного ХМЛ, или случаев предполагаемого ХМЛ, при котором филадельфийская хромосома не обнаруживается. У многих таких пациентов в действительности имеют место комплексные хромосомные аномалии, маскирующие транслокацию t(9;22), либо эта транслокация обнаруживается только при флюоресцентной гибридизации или ПЦР с обратной транскрипцией, но не при рутинном кариотипировании[11]. Для небольшой подгруппы пациентов с отсутствием молекулярных свидетельств присутствия гена BCR-ABL может быть поставлен диагноз недифференцированное миелодиспластическое/миелопролиферативное расстройство, так как оно, как правило, отличается от ХМЛ по клиническому течению[8].
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Nowell P.C. Discovery of the Philadelphia chromosome: a personal perspective (англ.) // Journal of Clinical Investigation (англ.)русск. : journal. — 2007. — Vol. 117, no. 8. — P. 2033—2035. — DOI:10.1172/JCI31771. — PMID 17671636.
- ↑ 1 2 3 Faderl S., Talpaz M., Estrov Z., Kantarjian H.M. Chronic myelogenous leukemia: biology and therapy (англ.) // Annals of Internal Medicine (англ.)русск.. — 1999. — Vol. 131, no. 3. — P. 207—219. — PMID 10428738.
- ↑ 1 2 3 Hehlmann R., Hochhaus A., Baccarani M; European LeukemiaNet. Chronic myeloid leukaemia (англ.) // The Lancet. — Elsevier, 2007. — Vol. 370, no. 9584. — P. 342—350. — DOI:10.1016/S0140-6736(07)61165-9. — PMID 17662883.
- ↑ 1 2 3 4 5 6 Tefferi A. Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F era (англ.) // Hematology Am Soc Hematol Educ Program : journal. — 2006. — Vol. 2006. — P. 240—245. — DOI:10.1182/asheducation-2006.1.240. — PMID 17124067.
- ↑ Kantarjian H., Dixon D., Keating M., Talpaz M., Walters R., McCredie K., Freireich E. Characteristics of accelerated disease in chronic myelogenous leukemia (англ.) // Cancer (англ.)русск. : journal. — Wiley-Blackwell (англ.)русск., 1988. — Vol. 61, no. 7. — P. 1441—1446. — DOI:10.1002/1097-0142(19880401)61:7<1441::AID-CNCR2820610727>3.0.CO;2-C. — PMID 3162181.
- ↑ Sokal J., Baccarani M., Russo D., Tura S. Staging and prognosis in chronic myelogenous leukemia (англ.) // Semin Hematol : journal. — 1988. — Vol. 25, no. 1. — P. 49—61. — PMID 3279515.
- ↑ 1 2 3 Tefferi A., Thiele J., Orazi A., Kvasnicka H.M., Barbui T., Hanson C.A., Barosi G., Verstovsek S., Birgegard G., Mesa R., Reilly J.T., Gisslinger H., Vannucchi A.M., Cervantes F., Finazzi G., Hoffman R., Gilliland D.G., Bloomfield C.D., Vardiman J.W. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert pane (англ.) // Blood (англ.)русск. : journal. — American Society of Hematology (англ.)русск., 2007. — Vol. 110, no. 4. — P. 1092—1097. — DOI:10.1182/blood-2007-04-083501. — PMID 17488875.
- ↑ Vardiman J., Harris N., Brunning R. The World Health Organization (WHO) classification of the myeloid neoplasms (англ.) // Blood (англ.)русск. : journal. — American Society of Hematology (англ.)русск., 2002. — Vol. 100, no. 7. — P. 2292—2302. — DOI:10.1182/blood-2002-04-1199. — PMID 12239137. Архивировано 6 ноября 2006 года.
- ↑ Karbasian Esfahani M., Morris E.L., Dutcher J.P., Wiernik P.H. Blastic phase of chronic myelogenous leukemia (неопр.) // Current Treatment Options in Oncology. — 2006. — Т. 7, № 3. — С. 189—199. — DOI:10.1007/s11864-006-0012-y. — PMID 16615875.
- ↑ Savage DG; Szydlo RM; Goldman J.M. Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period (англ.) // British Journal of Haematology (англ.)русск. : journal. — 1997. — Vol. 96, no. 1. — P. 111—116. — DOI:10.1046/j.1365-2141.1997.d01-1982.x. — PMID 9012696.
Острый промиелоцитарный лейкоз — Википедия
Острый промиелоцитарный лейкоз (ОПМЛ, ОПЛ) — это особый подвид острого миелоидного лейкоза (ОМЛ), злокачественного опухолевого заболевания белых кровяных клеток, по Франко-Американо-Британской классификации (ФАБ) относящийся к подтипу М3.[1] При ОПЛ наблюдается аномальное накопление в костном мозгу и в крови незрелых гранулоцитов, так называемых промиелоцитов. Эта форма ОМЛ характеризуется типичной хромосомной транслокацией, вовлекающей ген рецептора ретиноевой кислоты типа альфа (RARα или RARA) и ген белка острого промиелоцитарного лейкоза (PML), что приводит к образованию аномального фузионного онкопротеина PML-RARalpha и к неконтролируемому размножению мутантных промиелоцитов. Эта форма ОМЛ также отличается уникальным ответом на лечение полностью транс-ретиноевой кислотой и триоксидом мышьяка. Острый промиелоцитарный лейкоз был впервые описан и охарактеризован в 1957 году[2][3]французскими и норвежскими врачами-онкогематологами. В то время ОПЛ считался сверхострым и фатальным заболеванием, одной из самых неблагоприятных форм ОМЛ.[1] В настоящее время ОПЛ с классической транслокацией, напротив, считается одной из самых благоприятных и хорошо излечимых форм острого миелоидного лейкоза, с 12-летней выживаемостью без прогрессирования приблизительно 70 %.[1][4]
Симптомы ОПЛ в целом сходны с симптомами других форм ОМЛ. В частности, возможны следующие симптомы:[5]
Кровоточивость и кровотечения вследствие тромбоцитопении (пониженного содержания тромбоцитов в крови) может наблюдаться в следующих формах:
Острый промиелоцитарный лейкоз обычно характеризуется наличием реципрокной хромосомной транслокации между участком хромосомы 17, содержащим ген рецептора ретиноевой кислоты типа альфа, и участком хромосомы 15, содержащим ген так называемого белка острого промиелоцитарного лейкоза (PML). Эта транслокация приводит к образованию аномального онкогенного фузионного протеина PML-RARalpha. Эта транслокация обозначается как t(15;17)(q22;q21) и встречается в 95 % случаев ОПЛ. А ген рецептора RAR зависит от ретиноевой кислоты для регуляции транскрипции.[1]
Описаны при ПМЛ также 8 типов других редких хромосомных транслокаций, приводящих к формированию других аномальных фузионных онкопротеинов гена RARalpha с другими генами, в частности транслокация t(11;17), приводящая к слиянию гена RARalpha с геном так называемого «цинкового пальцеобразного белка промиелоцитарного лейкоза» (c геном PLZF, известным также как ген ZBTB16)[6], с геном нуклеофосмина (NPM1), с геном, ассоциированным с ядерным матриксом (NUMA1), с геном фактора 5b сигнальной трансдукции и активации транскрипции (STAT5B), с геном регуляторной субъединицы 1α протеинкиназы А (PRKAR1A), с геном фактора, взаимодействующего с белками PAPOLA и CPSF1 (FIP1L1), с геном корепрессора BCL-6 или с геном олигонуклетид/олигосахарид-связывающей последовательности 2A (OBFC2A, также известный как NABP1). Некоторые из этих хромосомных аномалий также являются чувствительными к лечению полностью транс-ретиноевой кислотой, либо их чувствительность к лечению полностью транс-ретиноевой кислотой неизвестна вследствие большой редкости этих хромосомных перестроек. Известно, что формы ОПЛ с хромосомными перестройками, приводящими к образованию аномальных фузионных онкопротеинов STAT5B/RARA и PLZF/RARA, резистентен к лечению полностью транс-ретиноевой кислотой и поэтому имеют худший прогноз.[1]
Слияние генов белков PML и RARA в результате транслокации t(15;17)(q22;q21) приводит к образованию аномального гибридного фузионного онкопротеина с изменёнными (необычными) внутриклеточными функциями. Этот аномальный онкопротеин с повышенным аффинитетом связывается с регуляторными участками внутриклеточной ДНК, блокируя транскрипцию важных генов, необходимых для процесса созревания и дифференцировки гранулоцитов. Он делает это при помощи усиленного взаимодействия молекул белка ядерного корепрессора (NCOR) и молекул гистон-деацетилазы (HDAC). Хотя хромосомная транслокация, включающая ген RARalpha, считается необходимой для инициации (начала) процесса лейкозогенеза, она сама по себе не достаточна для возникновения ПМЛ — требуется также приобретение этими клетками дополнительных мутаций.[1]
Мутация t(11;17), приводящая к образованию аномального фузионного онкопротеина RAR-α/PLZF приводит к развитию такого подтипа ОПЛ, который не чувствителен к стандартной терапии полностью транс-ретиноевой кислотой и/или триоксидом мышьяка и менее чувствителен, по сравнению с более частым вариантом RAR-α/PML, к стандартной химиотерапии ОМЛ, в частности к цитарабину и антрациклинам, таким, как даунорубицин, идарубицин, митоксантрон. Это приводит к худшему прогнозу в этой подгруппе пациентов.[1]
Острый промиелоцитарный лейкоз может быть отдифференцирован от других форм ОМЛ на основании патоморфологического исследования трепанобиоптата и аспирата костного мозга, а также крови, при котором может быть установлено, что бластные клетки по своему строению являются промиелоцитами. Дальнейшая диагностика требует цитогенетического исследования на характерные транслокации длинных плеч хромосом 17 и 15 или 17 и 11 — t(15,17) или t(11,17) и тестирования на наличие фузионных генов PML/RARA или PLZF/RARA, что может быть сделано при помощи полимеразной цепной реакции или флуоресцентной in situ гибридизации. Иногда могут встречаться скрытые транслокации, не обнаруживаемые при стандартном цитогенетическом исследовании. В подобных случаях полимеразная цепная реакция (ПЦР) критически необходима для подтверждения (верификации) диагноза.[1] Наличие в бластных клетках периферической крови большого количества телец Ауэра делает диагноз острого промиелоцитарного лейкоза очень вероятным, так как для бластных промиелоцитов очень характерно обилие телец Ауэра (по сравнению с более ранними бластными клетками типов M0, M1 и M2).
Индукционное лечение[править | править код]
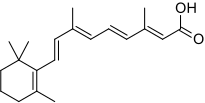 Полностью транс-ретиноевая кислота
Полностью транс-ретиноевая кислота
ОПЛ является уникальным среди острых миелоидных лейкозов по своей чувствительности к полностью транс-ретиноевой кислоте, кислой форме витамина А и к триоксиду мышьяка.[1] Лечение полностью транс-ретиноевой кислотой вызывает диссоциацию белкового комплекса RAR с белками NCOR (ядерным корепрессором) и HDACL (лиганд гистон-деацетилазы) и позволяет, напротив, связываться с рецептором RAR активаторам транскрипции и гистон-ацетилтрансферазам. Это, в свою очередь, позволяет запуститься процессу транскрипции нужных генов и процессу дифференцировки незрелых лейкозных промиелоцитов в зрелые гранулоциты. Таким образом, благодаря влиянию полностью транс-ретиноевой кислоты, «исправляется» аберрантная работа онкогенного транскрипционного фактора PML/RARalpha. При этом, в отличие от традиционной химиотерапии, полностью транс-ретиноевая кислота непосредственно не убивает злокачественные клетки. Вместо этого полностью транс-ретиноевая кислота вызывает их терминальную дифференцировку, после которой они подвергаются спонтанному апоптозу подобно здоровым гранулоцитам, у которых истёк отведённый им срок жизни (причём «в норме», до приобретения злокачественным клоном анти-апоптотических мутаций, апоптоз этих дифференцировавшихся под влиянием ATRA злокачественных клеток происходит гораздо быстрее, чем апоптоз здоровых гранулоцитов).[1] Полностью транс-ретиноевая кислота одна (в монотерапии) способна вызвать ремиссию при ОПЛ. Однако ремиссия, вызванная применением одной лишь ATRA, при ОПЛ, как правило, кратковременна в отсутствие одновременно проводимой «традиционной» химиотерапии и/или лечения триоксидом мышьяка, так как лейкозные клетки довольно быстро приобретают устойчивость к ATRA.[1] Начиная с 2013 года стандартом лечения ОПЛ является одновременное применение ATRA и триоксида мышьяка (плюс-минус традиционная химиотерапия).[7] До 2013 года стандартом лечения ОПЛ было одновременное применение ATRA и традиционной химиотерапии, основанной на применении антрациклинов (даунорубицин, доксорубицин, идарубицин или митоксантрон).
Ранее, до выявления того факта, что ОПЛ менее других разновидностей ОМЛ нуждается в традиционной химиотерапии и менее других разновидностей ОМЛ чувствителен к цитарабину, зато более других разновидностей чувствителен к антрациклинам, и что добавление цитарабина или других агентов не увеличивает процент полных ремиссий и общую выживаемость по сравнению с сочетанием «ATRA + антрациклин», зато более токсично, вместо схемы «ATRA + монотерапия антрациклином» часто применяли лечение «ATRA + та или иная схема традиционной противолейкозной химиотерапии» (например, ATRA + «7+3» или ATRA + ADE). Обе разновидности лечения (ATRA плюс триоксид мышьяка и ATRA плюс традиционная химиотерапия) приводят к примерно одинаковым результатам — около 90—95 % полных клинических и гематологических ремиссий. Но поскольку триоксид мышьяка менее токсичен и даёт меньше побочных эффектов, чем традиционная химиотерапия, и более специфичен в отношении клеток ОПЛ, то именно сочетание ATRA и триоксида мышьяка стало новым стандартом лечения ОПЛ.[4]
Лечение полностью транс-ретиноевой кислотой ассоциируется с уникальным побочным эффектом, ранее называвшимся «синдром ретиноевой кислоты» (более современное и корректное название «дифференциационный синдром» или «синдром дифференциации»)].[8] Дифференциационный синдром проявляется затруднением лёгочного дыхания (диспноэ), головной болью, лихорадкой (гипертермией), прибавкой массы тела, периферическими отеками, коагулопатией (кровоточивостью или тромбозами, ДВС-синдромом). Лечится дифференциационный синдром немедленным назначением глюкокортикоидов, например, преднизолона или дексаметазона, назначением гепарина для профилактики ДВС-синдрома и немедленным присоединением (или увеличением дозы) химиотерапии к уже проводящемуся лечению ATRA. При резко выраженном дифференциационном синдроме может потребоваться временная отмена или временное снижение дозы ATRA.[9] Этиологию дифференциационного синдрома связывают с массивным высвобождением цитокинов из массово дифференцирующихся и затем погибающих лейкозных промиелоцитов, что приводит к резкому повышению проницаемости капилляров (отеки, диспноэ и др.) и нарушениям в свертывающей системе крови.[9]
Моноклональное антитело гемтузумаб озогамицин (Милотарг) в комбинации с ATRA также было успешно использовано в лечении ОПЛ.[10] Однако оно было изъято с рынка США из-за подозрений в чрезмерной токсичности этого лекарства и отсутствии у него положительного влияния на общую и безрецидивную выживаемость больных с ОМЛ, отсутствии преимуществ перед традиционной химиотерапией. В Австралии, Канаде или Великобритании это лекарство никогда не продавалось.[10][11] Гемтузумаб озогамицин в комбинации с ATRA вызывал положительный ответ у приблизительно 84 % пациентов с ОПЛ, что сравнимо с цифрами, наблюдавшимися у пациентов, лечившихся традиционной комбинацией «ATRA плюс антрациклин».[10] Поскольку гемтузумаб озогамицин обладает меньшей кардиотоксичностью, чем антрациклины, такой вариант лечения мог бы быть более предпочтительным для пожилых больных с ОПЛ и больных с сопутствующими сердечно-сосудистыми заболеваниями.[10]
Консолидационное лечение[править | править код]
После достижения стойкой ремиссии стандартно рекомендуется 2 года поддерживающей терапии ATRA в сочетании с меркаптопурином и метотрексатом.[12] Значительный процент больных с ОПЛ рецидивирует без проведения поддерживающей терапии.[9] В европейском исследовании по ОПЛ, частота рецидивов в течение 2 лет среди тех, кто не получал поддерживающей химиотерапии, составляла 27 %, по сравнению с 11 % рецидивов у тех, которые поддерживающую химиотерапию получали.[13] Аналогично, в исследовании по ОПЛ, проведённом в США в 2000 году, общая 5-летняя выживаемость среди больных с ОПЛ, получавших поддерживающую терапию ATRA, составляла 61 %, по сравнению с 31 % среди тех, кто поддерживающую терапию ATRA не получал.[14]
Рецидив или рефрактерное заболевание[править | править код]
При рецидивах и рефрактерности ОПЛ в настоящее время изучается применение триоксида мышьяка. Были сообщения о достижении ремиссии при помощи монотерапии триоксидом мышьяка.[15] Исследования показали, что триоксид мышьяка вызывает реорганизацию ядерных телец и способствует деактивации и деградации мутантного PML-RAR онкопротеина.[16] Триоксид мышьяка также повышает активность каспазы, что вызывает апоптоз клеток.[17] Подобно ATRA, профилактическое применение триоксида мышьяка в поддерживающей терапии способно снизить риск рецидивов при ОПЛ, особенно у пациентов из группы высокого риска.[18] В Японии синтетический ретиноид, тамибаротен, лицензирован для применения при резистентном к ATRA ОПЛ.[19]
Помимо этого, при рецидивах и рефрактерности ОПЛ сохраняет своё значение и традиционная противолейкозная химиотерапия, в частности схемы «7+3», ADE, FLAG-подобные режимы и другие, а также высокодозная химиотерапия и аллогенная трансплантация гемопоэтических стволовых клеток.
Исследовательские агенты[править | править код]
Некоторые исследования показывают возможную потенциальную эффективность ингибиторов гистон-деацетилазы, таких, как вориностат или вальпроевая кислота, в лечении ОПЛ.[20][21][22]
Острый промиелоцитарный лейкоз представляет сравнительно редкую форму ОМЛ, всего около 10 — 12 % от всех случаев ОМЛ.[10] Средний возраст больных ОПЛ — приблизительно 30 — 40 лет,[23] что гораздо моложе, чем средний возраст больных с другими подтипами ОМЛ (70 лет). Частота ОПЛ выше среди пациентов латиноамериканского или южноевропейского происхождения.[24] ОПЛ может также возникнуть как вторичный лейкоз у больных, получавших лечение антрациклинами (например, доксорубицином, идарубицином, митоксантроном), алкилирующими агентами (например, циклофосфамидом) или ингибиторами топоизомеразы II (например, этопозидом), вследствие канцерогенности этих агентов. Большинство пациентов, получавших лечение этими агентами, у которых впоследствии развился ОПЛ, составляют больные с раком молочной железы в анамнезе.[25][26][27] У приблизительно 40 % пациентов с ОПЛ обнаруживаются, помимо t(15;17)(q22;q21), приводящей к появлению PML/RARalpha, дополнительные хромосомные аномалии в лейкозных клетках, такие, как трисомия 8-й хромосомы или изохромосома 17, но они, по-видимому, не оказывают существенного влияния на долгосрочный прогноз.[1]
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 Kotiah, SD; Besa, EC. Sarkodee-Adoo, C; Talavera, F; Sacher, RA; McKenna, R; Besa, EC: Acute Promyelocytic Leukemia (неопр.). Medscape Reference. WebMD (3 июня 2013). Дата обращения 14 января 2014.
- ↑ Tallman M.S., Altman J.K. Curative strategies in acute promyelocytic leukemia (неопр.) // Hematology Am Soc Hematol Educ Program. — 2008. — Т. 2008. — С. 391—399. — DOI:10.1182/asheducation-2008.1.391. — PMID 19074116.
- ↑ Hillestad, L.K. Acute promyelocytic leukemia (неопр.) // Acta Med Scand.. — 1957. — November (т. 159, № 3). — С. 189—194. — DOI:10.1111/j.0954-6820.1957.tb00124.x. — PMID 13508085.
- ↑ 1 2 Adès, L; Guerci, A; Raffoux, E; Sanz, M; Chevallier, P; Lapusan, S; Recher, C; Thomas, X; Rayon, C; Castaigne, S; Tournilhac, O; de Botton, S; Ifrah, N; Cahn JY; Solary E; Gardin, C; Fegeux, N; Bordessoule, D; Ferrant, A; Meyer-Monard, S; Vey, N; Dombret, H; Degos, L; Chevret, S; Fenaux, P; European APL Group. Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: the European APL Group experience (англ.) // Blood (англ.)русск. : journal. — American Society of Hematology (англ.)русск., 2010. — March (vol. 115, no. 9). — P. 1690—1696. — DOI:10.1182/blood-2009-07-233387. — PMID 20018913. (недоступная ссылка)
- ↑ Kotiah, SD; Besa, EC. Sarkodee-Adoo, C; Talavera, F; Sacher, RA; McKenna, R; Besa, EC: Acute Promyelocytic Leukemia Clinical Presentation (неопр.). Medscape Reference. WebMD (3 июня 2013). Дата обращения 14 января 2014.
- ↑ Chen Z., Brand N.J., et al. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia (англ.) // The EMBO Journal (англ.)русск. : journal. — 1993. — March (vol. 12, no. 3). — P. 1161—1167. — PMID 8384553.
- ↑ Francesco Lo-Coco, M.D., et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia (англ.) // The New England Journal of Medicine : journal. — 2013. — July (vol. 369, no. 2). — P. 111—121. — DOI:10.1056/NEJMoa1300874.
- ↑ Breccia, M; Latagliata, R; Carmosino, I; Cannella, L; Diverio, D; Guarini, A; De Propris, MS; Petti, MC; Avvisati, G; Cimino, G; Mandelli, F; Lo-Coco, F. Clinical and biological features of acute promyelocytic leukemia patients developing retinoic acid syndrome during induction treatment with all-trans retinoic acid and idarubicin (англ.) // Haematologica (англ.)русск. : journal. — 2008. — December (vol. 93, no. 12). — P. 1918—1920. — DOI:10.3324/haematol.13510. — PMID 18945746.
- ↑ 1 2 3 Kotiah, SD; Besa, EC. Sarkodee-Adoo, C; Talavera, F; Sacher, RA; McKenna, R; Besa, EC: Acute Promyelocytic Leukemia Treatment & Management (неопр.). Medscape Reference. WebMD (3 июня 2013). Дата обращения 14 января 2014.
- ↑ 1 2 3 4 5 Ravandi, F; Estey, EH; Appelbaum, FR; Lo-Coco, F; Schiffer, CA; Larson, RA; Burnett, AK; Kantarjian, H.M. Gemtuzumab Ozogamicin: Time to Resurrect? (англ.) // Journal of Clinical Oncology (англ.)русск.. — 2012. — November (vol. 30, no. 32). — P. 3921—3923. — DOI:10.1200/JCO.2012.43.0132. — PMID 22987091.
- ↑ Martindale: The Complete Drug Reference (неопр.). — Pharmaceutical Press (англ.)русск., 2011.
- ↑ Kotiah, SD. Anand, J; Braden, CD; Harris, JE: Acute Promyelocytic Leukema Treatment Protocols (неопр.). Medscape Reference. WebMD (28 октября 2013). Дата обращения 14 января 2014.
- ↑ Fenaux, P; Chastang, C; Chevret, S; Sanz, M; Dombret, H; Archimbaud, E; Fey, M; Rayon, C; Huguet, F; Sotto, JJ; Gardin, C; Makhoul, PC; Travade, P; Solary, E; Fegueux, N; Bordessoule, D; Miguel, JS; Link, H; Desablens, B; Stamatoullas, A; Deconinck, E; Maloisel, F; Castaigne, S; Preudhomme, C; Degos, L. A Randomized Comparison of All Transretinoic Acid (ATRA) Followed by Chemotherapy and ATRA Plus Chemotherapy and the Role of Maintenance Therapy in Newly Diagnosed Acute Promyelocytic Leukemia (англ.) // Blood (англ.)русск. : journal. — American Society of Hematology (англ.)русск., 1999. — August (vol. 94, no. 4). — P. 1192—1200. — PMID 10438706. (недоступная ссылка)
- ↑ Tallman, MS; Andersen, JW; Schiffer, CA; Appelbaum, FR; Feusner, JH; Woods, WG; Ogden, A; Weinstein, H; Shepherd, L; Willman, C; Bloomfield, CD; Rowe, JM; Wiernik, P.H. All-transretinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol (англ.) // Blood (англ.)русск. : journal. — American Society of Hematology (англ.)русск., 2002. — December (vol. 100, no. 13). — P. 4298—4302. — DOI:10.1182/blood-2002-02-0632. — PMID 12393590. (недоступная ссылка)
- ↑ Soignet S.L., Maslak P., Wang Z.G., et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide (англ.) // The New England Journal of Medicine : journal. — 1998. — November (vol. 339, no. 19). — P. 1341—1348. — DOI:10.1056/NEJM199811053391901. — PMID 9801394.
- ↑ Soignet,Complete Remission After Treatment of APL with Arsenic Trioxide 1998, 1346
- ↑ Soignet, 1998, 1347
- ↑ Arsenic Compound Improves Survival in Acute Promyelocytic Leukemia Patients . Sept 2007
- ↑ Miwako, I; Kagechika, H. Tamibarotene (неопр.) // Drugs Today (Barc). — 2007. — August (т. 43, № 8). — С. 563—568. — DOI:10.1358/dot.2007.43.8.1072615. — PMID 17925887.
- ↑ Martens, JH; Brinkman, AB; Simmer, F; Francoijs, KJ; Nebbioso, A; Ferrara, F; Altucci, L; Stunnenberg, H.G. PML-RARa/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia (англ.) // Cancer Cell : journal. — 2010. — February (vol. 17, no. 2). — P. 173—185. — DOI:10.1016/j.ccr.2009.12.042. — PMID 20159609. Архивировано 15 марта 2013 года.
- ↑ Leiva, M; Moretti, S; Soilihi, H; Pallavicini, I; Peres, L; Mercurio, C; Dal Zuffo, R; Minucci, S; de Thé, H. Valproic acid induces differentiation and transient tumor regression, but spares leukemia-initiating activity in mouse models of APL (англ.) // Leukemia : journal. — 2012. — July (vol. 26, no. 7). — P. 1630—1637. — DOI:10.1038/leu.2012.39. — PMID 22333881.
- ↑ Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia (англ.) // He, LZ; Tolentino, T; Grayson, P; Zhong, S; Warrell, RP Jr; Rifkind, RA; Marks, PA; Richon, VM; Pandolfi, PP : journal. — Vol. 108, no. 9. — P. 1321—1330. — DOI:10.1172/JCI11537. — PMID 11696577.
- ↑ Schiffer, CA; Stone, R. M. Chapter 124: Acute Myeloid Leukemia in Adults // Holland-Frei Cancer Medicine (неопр.) / Bast, RC; Kufe, DW; Pollock, R. E.. — 5th. — Hamilton, ON: BC Decker, 2000.
- ↑ Douer, D; Santillana, S; Ramezani, L; Samanez, C; Slovak, ML; Lee, MS; Watkins, K; Williams, T; Vallejos, C. Acute promyelocytic leukaemia in patients originating in Latin America and is associated with an increased frequency of the bcr1 subtype of the PML/RARalpha fusion gene (англ.) // British Journal of Haematology (англ.)русск. : journal. — 2003. — August (vol. 122, no. 4). — P. 563—570. — DOI:10.1046/j.1365-2141.2003.04480.x. — PMID 12899711.
- ↑ Ravandi, F. Therapy-related acute promyelocytic leukemia (англ.) // Haematologica (англ.)русск.. — 2011. — April (vol. 96, no. 4). — P. 493—495. — DOI:10.3324/haematol.2011.041970. — PMID 21454880.
- ↑ Elliott, MA; Letendre, L; Tefferi, A; Hogan, WJ; Hook, C; Kaufmann, SH; Pruthi, RK; Pardanani, A; Begna, KH; Ashrani, AA; Wolanskyj, AP; Al-Kali, A; Litzow, M.R. Therapy-related acute promyelocytic leukemia: observations relating to APL pathogenesis and therapy (англ.) // European Journal of Haematology : journal. — 2012. — March (vol. 88, no. 3). — P. 237—243. — DOI:10.1111/j.1600-0609.2011.01727.x. — PMID 22023492.
- ↑ Rashidi, A; Fisher, S.I. Therapy-related acute promyelocytic leukemia: a systematic review (англ.) // Medical Oncology : journal. — 2013. — Vol. 30, no. 3. — P. 625. — DOI:10.1007/s12032-013-0625-5. — PMID 23771799.
Острые миелобластные лейкозы — Википедия
Материал из Википедии — свободной энциклопедии
Острые миелобластные лейкозы — группа острых миелоидных лейкозов со злокачественным ростом миелобластов.
Франко-американско-британская классификация[править | править код]
По франко-американо-британской (FAB) классификации, к острым миелобластным лейкозам относятся восемь типов миелоидных лейкозов — минимально-дифференцированный острый миелобластный лейкоз (M0), острый миелобластный лейкоз без признаков созревания (M1), острый миелобластный лейкоз с признаками созревания (М2), промиелоцитарный, или острый промиелоцитарный лейкоз (ОПЛ) (М3), острый миеломоноцитарный лейкоз (М4), миеломоноцитарный сочетанный с эозинофилией костного мозга (М4ео), острый монобластный лейкоз (M5a) или острый моноцитарный лейкоз (M5b), острые эритроидные лейкозы, включая эритроцитарный лейкоз (M6a) и очень редкий чистый эритроидный лейкоз (M6b), острый мегакариобластный лейкоз (М7) и острый базофильный лейкоз (М8).[1]
Описываются наблюдения развития острого миелобластного лейкоза у людей, подвергшихся радиационному воздействию, контактирующих с бензолом (кожевенная индустрия в Турции, производство синтетических клеев и др.), принимавших цитостатические препараты, а также у страдающих наследственными заболеваниями — синдромом Дауна, анемией Фанкони, синдромом Блума.
Опухолевые клетки имеют типичные для миелобластов цитохимические маркеры: ШИК-положительную диффузно окрашенную цитоплазму, содержат липиды, пероксидазу, эстеразы. Опухолевые клетки инфильтрируют костный мозг, приобретающий макроскопически пиоидный вид, селезёнку, печень, лимфатические узлы, слизистую оболочку желудочно-кишечного тракта.
Инфильтрация тканей опухолевыми клетками сопровождается язвенно-некротическими и геморрагическими осложнениями. В 1/3 случаев лейкозные инфильтраты обнаруживаются в легких («лейкозный пневмонит»), в 1/4 — в оболочках мозга («лейкозный менингит»). По иммунологическим фенотипам выделяют 6 вариантов заболевания.
Больные умирают от кровоизлияния в головной мозг, желудочно-кишечных кровотечений и инфекционных осложнений.
Лечение цитостатиками изменило проявления заболевания, удлинило жизнь больным.
За последние 10 лет благодаря применению новых подходов к лечению детей с ОМЛ с включением новых химиотерапевтических агентов и ТКМ удалось увеличить выживаемость больных до 50%. Источник
- Патологическая анатомия. Курс лекций. Под ред. В. В. Серова, М. А. Пальцева. — М.: Медицина, 1998
- Шулутко Б. И., Макаренко С. В. Стандарты диагностики и лечения внутренних болезней. 3-е изд. СПб.: «Элби-СПБ», 2005
Гемобластозы
Лейкозы
Острые лейкозы
Лейкоз Википедия
| Лейкоз | |
|---|---|
 Микропрепарат костного мозга больного острой В-лимфоцитарной лейкобластной лейкемией | |
| МКБ-10 | C9191.-C9595. |
| МКБ-10-КМ | C95.90, C95 и C95.9 |
| МКБ-9 | 208.9208.9 |
| МКБ-9-КМ | 208[1][2], 208.9[1][2], 207.8[2], 208.80[2], 207[2], 208.90[2], 207.80[2] и 208.8[2] |
| МКБ-О | 9800-9940 |
| DiseasesDB | 7431 |
| MedlinePlus | 001299 |
| MeSH | D007938 |
Лейко́з (греч. λευκός — белый) (лейкеми́я (греч. λευκός — белый и греч. αἷμα — кровь), алейкеми́я[3], белокровие, рак крови) — клональное злокачественное (неопластическое) заболевание кроветворной системы. К лейкозам относится обширная группа заболеваний, различных по своей этиологии. При лейкозах злокачественный клон может происходить как из незрелых гемопоэтических клеток костного мозга, так и из созревающих и зрелых клеток крови.
Лейкоз выражается системной диффузной гиперплазией всего клеточного аппарата, продуцирующего лейкоциты, независимо от того, сопровождаются ли они белокровием или протекают без него.[4]
Гемобластозы — Википедия
Материал из Википедии — свободной энциклопедии
Гемобластозы (лат. haemoblastosis; др.-греч. αἷμα «кровь» + βλαστός росток, зародыш + -osis) — опухолевые (неопластические) заболевания кроветворной и лимфатической ткани.
Гемобластозы подразделяют на системные заболевания — лейкозы, а также регионарные — лимфомы.
Отличия между лейкозами и лимфомами заключаются не только в наличии или отсутствии системности поражения. В терминальной стадии лимфомы дают обширные метастазы, в том числе и в костный мозг. Но при лейкозах костный мозг поражается первично, а при лимфомах — вторично в результате метастазирования. При лейкозах опухолевые клетки, как правило, обнаруживаются в крови, поэтому в литературе используется термин для обозначения лейкозов, предложенный ещё Р. Вирховом — «лейкемия».
Опухоли кроветворной и лимфоидной ткани в числе пяти самых распространенных опухолей человека. Среди опухолей детей первых 5 лет жизни на их долю приходится 30 % случаев.
Факторы, способствующие возникновению гемобластозов.
- Разнообразные мутагенные факторы экзогенного и эндогенного происхождения.
- Наследственность. Её роль подтверждается частым развитием лейкозов у людей с наследственными заболеваниями со спонтанными разрывами хромосом (болезнь Дауна, Блума, анемия Фанкони), с нерасхождением половых хромосом (болезни Клайнфельтера, Тернера), а также существованием «лейкозных семей». Нередко лейкозы развиваются у пациентов с наследственными дефектами иммунитета (атаксия-телеангиэктазия, или синдром Луи-Бар, синдром Вискотта-Олдриджа, болезнь Братона).
- Ионизирующая радиация. Её роль доказывается наблюдениями за пациентами, заболевшими лейкозами и лимфомами через определенное время после атомной бомбардировки Японии, аварий на АЭС, ядерных испытаний. Описаны случаи заболеваний у людей, получавших радиотерапию, а также у врачей-радиологов. Известен цитогенетический маркер радиационного поражения — кольцевидная хромосома. Достоверно установлена связь между радиационным поражением и развитием острого и хронического миелолейкоза, острого эритромиелолейкоза и острого лимфобластного лейкоза у детей.
- Химические канцерогены. Их роль доказывается данными экспериментов, наблюдениями за пациентами, работавшими на вредных предприятиях с использованием бензола, а также за больными, получавшими цитостатическую терапию по поводу других онкологических заболеваний. Использование таких цитостатических препаратов, как мелфалан, азатиоприн, лейкеран, миелосан, антибиотика левомицетина, может приводить к возникновению острого и хронического миелолейкоза, острого миеломонобластного лейкоза и эритромиелоза.
- Вирусы. В развитии гемобластозов человека доказано участие двух вирусов: вируса Эпштейна — Барр (африканская лимфома Беркитта) и T-лимфоцитарного вируса лейкоза человека первого типа (T-клеточная лимфома и клеточные лейкозы). Имеются экспериментальные данные, указывающие прямое канцерогенное действие вирусов на гемопоэтические клетки посредством вирусных онкогенов. Однако, в большинстве ситуаций внедрение вирусов в клетку вызывает лишь иммортализацию (бессмертие) последней, на фоне которой возникают дополнительные перестройки генома, ведущие к злокачественной трансформации (многоступенчатый канцерогенез).
Всё множество этиологических факторов, воздействуя на стволовые и полустволовые гемопоэтические клетки, приводят к одинаковым результатам — злокачественной трансформации.
Например, при лимфоме Бёркитта происходит реципрокная транслокация между хромосомами 8 и 14q32. Клеточный онкоген c-myc из хромосомы 8 перемещается на хромосому 14 и попадает в зону действия генов, регулирующих синтез тяжёлых цепей иммуноглобулинов. Описанные изменения сочетаются также с точечной мутацией N-ras.
При хроническом миелолейкозе нередко встречается филадельфийская хромосома, образовавшаяся в результате реципрокной транслокации между хромосомами 9 и 22. Образуется новый ген-гибрид c-abl-bcr, белковый продукт которого обладает тирозинкиназной активностью. Онкогены обычно встраиваются в разрывы хромосом. Так, при B-лимфоцитарных лимфомах и лейкозах происходят разрывы в хромосоме 14 в локусе 32q, где локализуются гены тяжёлых цепей иммуноглобулинов. При T-лимфоцитарных лейкозах и лимфомах — в локусе 11q гена α-цепей рецепторов T-лимфоцитов.
Развитие гемобластозов начинается с малигнизации одной стволовой или полустволовой клетки, дающей пул опухолевых клеток. Это означает, что все гемобластозы имеют моноклоновое происхождение. Моноклоновость происхождения подтверждается экспериментальными и клиническими данными по обнаружению во всех опухолевых клетках одного и того же больного клоновой метки — хромосомной или изоферментной. Например, во всех опухолевых клетках при хроническом миелолейкозе присутствует филадельфийская хромосома.
Стволовые клетки составляют примерно 0,01-0,001 % всей популяции костномозговых клеток. Рост и дифференцировка стволовых и полустволовых клеток-предшественниц управляются факторами роста и стромальным микроокружением. Это подтверждается экспериментами с клеточными культурами, где рост и дифференцировка клеток происходят только в присутствии факторов роста или стромальных клеток.
Многочисленные работы по изучению морфологии и клеточной кинетики гемобластозов (в первую очередь лейкозов) показали, что при их развитии происходит не только малигнизация на уровне стволовых и полустволовых клеток-предшественниц, но также развивается блок дифференцировки в пуле опухолевых клеток.
- Краткая Медицинская Энциклопедия — Петровский Б.В., 1984
- Патологическая анатомия. Курс лекций. Под ред. В. В. Серова, М. А. Пальцева. — М.: Медицина, 1998
Вирус лейкоза кошачьих — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 20 сентября 2018; проверки требует 1 правка. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 20 сентября 2018; проверки требует 1 правка.| Вирус лейкоза кошачьих | |||
|---|---|---|---|
промежуточные ранги
| |||
Feline leukemia virus | |||
VI: оцРНК-ОТ-вирусы | |||
Вирусная лейкимия кошачьих[2] (ВЛК, лат. Feline leukemia virus, FeLV) — вид ретровирусов, инфицирующих представителей семейства кошачьих. Вызывает у кошек заболевание, похожее на рак, являющееся вирусной формой лейкоза. Признаётся очень опасной инфекцией, часто приводящей к летальному исходу[3].
Вирус лейкоза FeLV был впервые описан в 1964 году учёным Уильямом Джарретом (W. Jarrett et al., Nature 202:566) в университете Глазго. Имеет диаметр 100—110 нм и сферическую форму.
Существует четыре типа вируса: A, B, C и Т, которые также имеют несколько подтипов.
Территориально вирус распространён повсеместно, но неравномерно.
Наиболее восприимчивыми к инфекции являются котята в возрасте 4 месяцев. Источником инфекции являются стареющие кошки. Вирус находится в слюне, выделениях из носа.
Вирус часто вызывает снижение иммунитета, которое приводит к анемии и лейкопении.
Для профилактики заболеваний рекомендуется вакцинация. Первая вакцинация проводится на 9-ой неделе после рождения, а через 2—4 недели повторная. В дальнейшем рекомендуется ежегодная вакцинация.
- «Болести на котката», издателство «АРИА» 1999 г., стр. 297—300.
- Золототрубов А. П. Эпизоотология и профилактика ретровирусных инфекций кошек. (недоступная ссылка с 18-04-2017 [1000 дней]) Ветеринарная патология, 3, 2007
- Золототрубов А. П., Федосов Д. В. Рентгенологический метод в диагностике ретровирусных инфекций кошек. (недоступная ссылка с 18-04-2017 [1000 дней]) Ветеринарная патология, 4, 2008
- Шахов А. Г., Золототрубов А. П., Федосов Д. В. Методические рекомендации по диагностике, терапии и профилактике ретровирусных инфекций кошек. 2008 г.
- Федосов Д. В., Золототрубов А. П. Пара синтетических олигонуклеотидов-праймеров, используемых для выявления днк провируса лейкоза кошек эндогенного и экзогенного типа, и условия полимеразной цепной реакции с их использованием. Патент на изобретение № 2317329
- Золототрубов А. П., Федосов Д. В. Молекулярно-генетичекий метод в диагностике лейкоза кошек // Материалы XII международного конгресса по болезням мелких домашних животных, Москва. — 2004. — С. 79—80.
- Золототрубов А. П., Федосов Д. В., Гребенщиков А. В. Морфологические изменения в органах и тканях кошек инфицированных вирусом лейкоза // Практик. 2005. № 9—10. С. 74—79.
- Золототрубов А. П., Федосов Д. В., Востроилова Г. А., Жаркой Б. Л. Новое в терапии лейкоза кошек.
- Федосов Д. В., Золототрубов А. П. Изучение функциональной активности лейкоцитов у ВЛК-инфицированных кошек.
- Непоклонова И. В., Лунина Н. А., Ткачёв А. В., Логинов Н. В. Вирусная лейкемия кошек: особенности течения инфекционного процесса.
- Лежандр А. М. Вирус лейкемии кошек // Российский ветеринарный журнал. — № 1. — 2005. — С. 36—38.
- Ланор О., Фамоз Ф. Медиастинальная лимфосаркома и неврологические проявления у кошки // Ветеринар. — № 3. — 2005. — С. 4—7.
- J.M. Coffin, S.H. Hughes, H.E. Varmus. Retroviruses. CSHL Press. 1997.
- I.G. Winkler, M. L^helt, and R.L.P Flower. Epidemiology of Feline Foamy Virus and Feline Immunodeficiency Virus Infections in Domestic and Feral Cats: a Seroepidemiological Study // Journal of Clinical Microbiology September 1999, Vol. 37, No. 9. P 2848—2851.
- Z. Knotek, P.H. AJ^v6, M. Svoboda, M. Toman and V. Ramka Epidemiology of Feline Leukaemia and Feline Immunodeficiency Virus Infections in the Czech Republic // Journal of Veterinary Medicine, Series B. 1999. Volume 46. P. 665.