Синдром Альпорта — причины, симптомы, диагностика и лечение

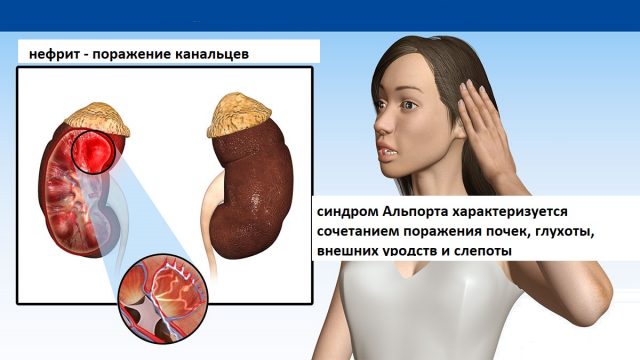
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.

Синдром Альпорта
Причины
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН. У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам. Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
Наследственный нефрит (синдром Альпорта) у детей : причины, симптомы, диагностика, лечение
Причины синдрома Альпорта
Генетическая основа болезни — мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики наследственного нефрита.
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена. У большинства больных наследственным нефритом в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения; имеют значение родственные браки между людьми, имеющими одного или более предков, так как в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У детей чаще различают три варианта наследственного нефрита: синдром Альпорта, наследственный нефрит без тугоухости и семейная доброкачественная гематурия.
Синдром Альпорта — наследственный нефрит с поражением слуха. В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома Альпорта расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
Генетической основой развития наследственного нефрита являются мутации в генах альфа-цепей коллагена IV типа. Известно шесть а-цепей IV коллагена Г типа: гены а5- и а6-цепей (Соl4A5 и Соl4A5) находятся на длинном плече Х-хромосомы в зоне 21-22q; гены а3- и а4-цепей (Соl4A3 и Соl4A4) — на 2-й хромосоме; гены a1- и a2-цепей (Соl4A1 и Соl4A2) — на 13-й хромосоме.
В большинстве случаев (80-85%) выявляется Х-сцепленный тип наследования заболевания, связанный с повреждениями гена Соl4A5 в результате делеции, точечных мутаций или нарушений сплайсинга. В настоящее время найдено более 200 мутаций гена Соl4A5, ответственных за нарушение синтеза а5-цепей коллагена IV типа. При этом типе наследования заболевание проявляется у детей обоего пола, но у мальчиков протекает тяжелее.
Мутации в локусах генов Соl4A3 и Соl4A4, ответственных за синтез а3- и а4 — цепей коллагена IV типа, наследуются аутосомно. По данным исследований аутосомно-доминантныи тип наследования отмечается в 16% случаев наследственного нефрита, аутосомно-рецессивный — у 6% больных. Изестно около 10 вариантов мутаций генов Соl4A3 и Соl4A4.
Результатом мутаций является нарушение процессов сборки коллагена IV типа, приводящее к нарушению его структуры. Коллаген IV типа является одним из основных компонентов гломерулярной базальной мембраны, кохлеарного аппарата и хрусталика глаза, патология которых будет выявляться в клинике наследственного нефрита.
Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV) и одной а2-цепи (IV), а также содержит а3, а4, а5-цепи. Наиболее часто при Х-сцепленном наследовании мутация гена Соl4A5 сопровождается отсутствием а3-, а4-, а5- и а6 цепей в структуре коллагена IV типа, а количество о1- и а2-цепей в гломерулярной базальной мембране возрастает. Механизм этого феномена неясен, предполагается, что причиной являются посттранскрипционные изменения мРНК.
Отсутствие а3-, а4- и а5-цепей в структуре IV типа коллагена базальных мембран клубочков приводит к их истончению и ломкости на ранних стадиях синдрома Альпорта, что клинически проявляется чаще гематурией (реже гематурией с протеинурией или только протеинурией), снижением слуха и лентиконусом. Дальнейшее прогрессирование заболевания приводит к утолщению и нарушению проницаемости базальных мембран на поздних стадиях заболевания, с разрастанием в них коллагена V и VI типов, проявляющихся в нарастании протеинурии и снижении почечных функций.
Характер мутации, лежащей в основе наследственного нефрита, во многом определяет его фенотипическое проявление. При делеции Х-хромосомы с одновременной мутацией генов Соl4A5 и Соl4A6, ответственных за синтез а5- и а6-цепей коллагена IV типа, синдром Альпорта сочетается с лейомиоматозом пищевода и половых органов. По данным исследований при мутации гена Соl4A5, связанной с делецией, отмечаются большая тяжесть патологического процесса, сочетание почечного поражения с экстраренальными проявлениями и ранним развитием хронической почечной недостаточности, по сравнению сточечной мутацией этого гена.
Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие электронно-плотных гранул. Поражение гломерул может быть неоднородным у одного и того же больного, от минимального фокального поражения мезангия до гломерулосклероза. Гломерулит при синдроме Альпорта всегда носит иммунонегативный характер, что отличает его от гломерулонефрита. Характерны развитие атрофии канальцев, лимфогистиоцитарная инфильтрация, наличие «пенистых клеток» с включениями липидов — липофагами. При прогрессировании заболевания выявляется утолщение и выраженная деструкция базальных мембран клубочков.
Выявляются определенные сдвиги в состоянии имунной системы. У больных наследственным нефритом отмечено снижение уровня Ig A и склонность к повышению концентрации IgМ в крови, уровень IgG может быть повышен на ранних стадиях развития заболевания и снижаться на поздних сроках. Возможно, повышение концентрации IgM и G является своеобразной компенсаторной реакцией в ответ на дефицит IgA.
Функциональная активность системы Т-лимфоцитов снижена; отмечается избирательное снижение В-лимфоцитов, ответственных за синтез Ig A, нарушается фагоцитарное звено иммунитета, в основном за счет нарушения процессов хемотаксиса и внутриклеточного переваривания в нейтрофилах
При исследовании биоптата почек у больных с синдромом Альпорта по данным электронной микроскопии наблюдаются ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях наследственного нефрита дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при наследственном нефрите — расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
 [8]
[8]
Синдром Альпорта у детей — симптомы и лечение
Генетически детерминированная неиммунная гломерулопатия, протекающая с гематурией, прогрессирующим снижением почечных функций — это синдром Альпорта или наследственный нефрит. Он проявляется комплексом патологий: наличием нефрита с гематурией, ухудшением слуха и патологией зрения. В этой статье мы расскажем вам про основные причины и симптомы синдрома, а также о том, как проводится его лечение у ребенка.
Причины синдрома Альпорта у детей
Согласно данным эпидемиологических исследований, проведенных в 13 регионах России, это заболевание встречается с частотой 17 на 100 000 детского населения [Игнатова М. С, 1999].
Этиология синдрома Альпорта
Генетическая основа заболевания – мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики заболевания [Цаликова Ф. Д. и др., 1995].
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена.
У большинства больных синдромом Альпорта, в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения, имеют значение родственные браки между людьми, имеющими одного или более предков, т.к. в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей [Фокеева В. В. и др., 1988]. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У малышей чаще различают три варианта синдрома Альпорта:
- непосредственно сам синдром,
- наследственный нефрит без тугоухости,
- семейная доброкачественная гематурия.
Патогенез синдрома Альпорта
В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
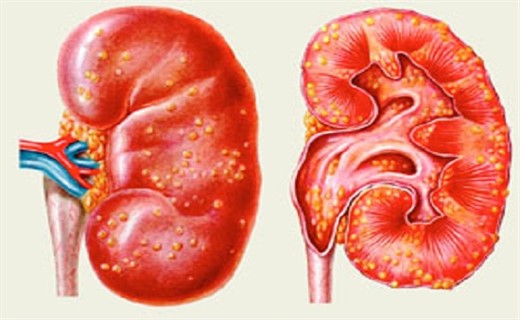
При исследовании биоптата почек по данным электронной микроскопии наблюдаются такие симптомы: ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях заболевания дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при данной болезни – расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
По каким симптомам узнать синдром Альпорта у детей?
Первые симптомы заболевания в виде изолированного мочевого синдрома чаще выявляются у малышей первых трех лет жизни. В большинстве случаев заболевание обнаруживается случайно. Мочевой синдром выявляется при профилактическом обследовании ребенка, перед поступлением в детское учреждение или во время ОРВИ. В случае появления патологии в моче во время ОРВИ при синдроме в отличие от приобретенного гломерулонефрита отсутствует латентный период.
Как проявляется синдром Альпорта в начальной стадии?
В начальной стадии самочувствие ребенка страдает мало, симптомы не явно выражены, проводится лечение согласно рекомендациям врача. Характерной особенностью является упорство и стойкость мочевого синдрома. Одним из основных признаков является гематурия различной степени выраженности, наблюдаемая в 100% случаев. Усиление степени гематурии отмечается во время или после инфекций дыхательных путей, физической нагрузки или после профилактических прививок. Протеинурия в большинстве случаев не превышает 1 г/сут, в начале заболевания может быть непостоянной, по мере прогрессирования процесса протеинурия нарастает. Периодически в мочевом осадке может присутствовать лейкоцитурия с преобладанием лимфоцитов, что связывают с развитием интерстициальных изменений.
В дальнейшем происходит нарушение парциальных функций почек, ухудшение общего состояния больного: появляются интоксикация, мышечная слабость, артериальная гипотония, часто нарушение слуха (особенно у мальчиков), иногда нарушение зрения. Интоксикация проявляется бледностью, утомляемостью, головными болями.
Снижение слуха — симптом появления синдрома Альпорта
В начальной стадии болезни снижение слуха в большинстве случаев выявляется только с помощью аудиографии. Снижение слуха может возникнуть в различные периоды детства, однако чаще всего тугоухость диагностируется в возрасте 6 — 10 лет. Начинается с высоких частот, достигая значительной степени при воздушном и костном проведении, переходя из звукопроводящей в звуковоспринимающую тугоухость. Снижение слуха может быть одним из первых симптомов заболевания и может предшествовать мочевому синдрому.
Снижение зрения — симптом синдрома Альпорта
В 20% случаев у больных отмечаются изменения со стороны органов зрения. Наиболее часто выявляются аномалии со стороны хрусталика: сферофокия, лентиконус передний, задний или смешанный, разнообразные катаракты. В семьях с с этим заболеванием наблюдается значительная частота миопии. Ряд исследователей постоянно в этих семьях отмечают билатеральные перимакулярные изменения в виде ярких беловатых или желтоватых грануляций в области желтого тела. Они считают этот признак постоянным симптомом, который имеет высокую диагностическую ценность при этом заболевании. К. S. Chugh и соавт. (1993) при офтальмологическом исследовании выявили у больных снижение остроты зрения в 66,7% случаев, передний лентиконус – в 37,8%, пятна на сетчатке – в 22,2%, катаракту – в 20%, кератоконус – в 6,7%.
Особенности синдрома Альпорта у детей
У части детей, особенно при формировании почечной недостаточности, отмечают существенное отставание в физическом развитии. По мере прогрессирования почечной недостаточности развивается артериальная гипертензия. У ребенка ее симптомы чаще выявляются в подростковом периоде и в более старших возрастных группах. При диагностировании лечение проводят незамедлительно.
Характерно наличие у больных с синдромом Альпорта разнообразных (более 5 — 7) стигм соединительнотканного дизэмбриогенеза [Фокеева В. В., 1989]. Среди соединительнотканных стигм у больных наиболее часто встречаются гипертелоризм глаз, высокое нёбо, аномалии прикуса, аномальная форма ушных раковин, искривление мизинца на руках, «сандалевидная щель» на стопах. Для заболевания характерны такие симптомы: однотипность стигм дизэмбриогенеза внутри семьи, а также высокая частота их распространения у родственников пробандов, по линии которых передается болезнь.
Клинические рекомендации о синдроме Альпорта
На ранних этапах болезни выявляется изолированное снижение парциальных функций почек: транспорта аминокислот, электролитов, концентрационной функции, ацидогенеза, в дальнейшем изменения касаются функционального состояния как проксимального, так и дистального отдела нефрона и носят характер сочетанных парциальных нарушений. Снижение клубочковой фильтрации наступает позднее, чаще в подростковом периоде. По мере прогрессирования развивается анемизация.
Таким образом, характерна стадийность течения заболевания: сначала латентная стадия или скрытых клинических симптомов, проявляющаяся минимальными изменениями мочевого синдрома, затем наступает постепенная декомпенсация процесса со снижением почечных функций с манифестными клиническими симптомами (интоксикация, астенизация, отставание в развитии, анемизация). Клинические симптомы появляются обычно вне зависимости от наслоения воспалительной реакции.
Синдром может манифестовать в разные возрастные периоды, что зависит от действия гена, который до определенного времени находится в репрессированном состоянии.
Как диагностируют синдром Альпорта у детей?
Предложены следующие критерии:
- Наличие в каждой семье не менее двух больных нефропатией,
- Гематурия как ведущий симптом нефропатии у пробанда,
- Наличие тугоухости хотя бы у одного из членов семьи,
- Развитие ХПН у одного родственника и более.
При диагностике разнообразных наследственных и врожденных заболеваний большое место принадлежит комплексному подходу к обследованию и прежде всего обращение внимания на данные, получаемые при составлении родословной ребенка. Диагноз синдрома Альпорта считается правомочным в случаях обнаружения у больного 3 из 4 типичных признаков: наличие в семье гематурии и хронической почечной недостаточности, присутствие у больного нейросенсорной тугоухости, патологии зрения, обнаружение при электронно-микроскопической характеристике биоптата признаков расщепления гломерулярной базальной мембраны с изменением ее толщины и неравномерностью контуров [Игнатова М. С, 1996].
Клинико-генетические методы исследования синдрома Альпорта
Прежде чем начнется лечение, проводится обследование больного, которое должно включать клинико-генетические методы исследования, направленное изучение анамнеза заболевания, общий осмотр больного с учетом диагностически значимых критериев.
- В стадии компенсации уловить патологию можно лишь ориентируясь на такие синдромы, как наличие наследственной отягощенности, гипотонии, множественных стигм дизэмбриогенеза, изменения мочевого синдрома.
- В стадии декомпенсации возможно появление эстраренальных симптомов, таких как выраженная интоксикация, астенизация, отставание в физическом развитии, анемизация, проявляющиеся и усиливающиеся с постепенным снижением почечных функций. У большинства больных при снижении почечных функций наблюдается: снижение функции ацидо- и аминогенеза, у 50% больных отмечают значительное снижение секреторной функции почек, ограничение пределов колебания оптической плотности мочи, нарушение ритма фильтрации, а затем и снижение клубочковой фильтрации.
- Стадия хронической почечной недостаточности диагностируется при наличии у больных в течение 3-6 мес. и более повышенного уровня мочевины в сыворотке крови (более 0,35 г/л), снижение клубочковой фильтрации до 25% от нормы.
Дифференциальная диагностика синдрома Альпорта
Ее приходится проводить с гематурической формой приобретенного гломерулонефрита. Приобретенный гломерулонефрит имеет чаще острое начало, период 2 — 3 недели после перенесенной инфекции, экстраренальные признаки, в том числе гипертензию с первых дней (при синдроме Альпорта, напротив, гипотония), снижение клубочковой фильтрации в начале заболевания, отсутствие нарушения парциальных канальцевых функций, тогда как при наследственном они присутствуют. Приобретенный гломерулонефрит протекает с более выраженной гематурией и протеинурией, с увеличенной СОЭ. Диагностическое значение имеют типичные изменения гломерулярной базальной мембраны, свойственные синдрому. Лечение должно быть начато оперативно.
Дифференциальная диагностика от дисметаболической нефропатии проводится с хронической почечной недостаточностью, в семье клинически выявляются неоднотипные болезни почек, а может быть спектр нефропатии от пиелонефрита до мочекаменной болезни. Часто присутствуют жалобы на боли в животе и периодически при мочеиспускании, в осадке мочи – оксалаты.
При подозрении на заболевание, больного необходимо направить для уточнения диагноза в специализированное нефрологическое отделение.

Как лечить синдром Альпорта у детей?
В режиме лечения предусматривается ограничение от больших физических нагрузок, пребывание на свежем воздухе. В период, когда проводится лечение показана полноценная диета, с достаточным содержанием полноценных белков, жиров и углеводов с учетом функции почек. Большое значение имеет выявление и санация хронических очагов инфекции. Из лекарственных средств используются АТФ, кокарбоксилаза, пиридоксин (до 50 мг/сут.), витамин В5, карнитина хлорид. Курсы проводят 2 — 3 раза в год. При гематурии назначается фитотерапия – крапива двудомная, сок черноплодной рябины, тысячелистник.
В зарубежной и отечественной литературе имеются сообщения о лечении преднизолоном и использовании цитостатиков. Однако об эффекте судить трудно.
Лечение синдрома Альпорта
При хронической почечной недостаточности применяются гемодиализ и трансплантация почек.
М. С. Игнатова (1999) считает, что основным методом при развитии ХПН является своевременное проведение трансплантации почки, которое возможно и без предварительного экстракорпорального диализа. Актуальной является проблема использования методов генной инженерии.
Необходима преемственность в постоянном наблюдении за больными и передача детей педиатром непосредственно терапевту-нефрологу. Диспансерное наблюдение осуществляется на протяжении всей жизни больного.
Теперь вы знаете основные симптомы и способы лечения синдрома Альпорта у детей. Здоровья вашему малышу!
Синдром Альпорта у детей: лечение, диагностика
Болезнь под название Синдром Альпорта передается наследственным путем и проявляется в симптоматике снижения функций почек совместно с сопутствующими нарушениями зрения и слуха. По статистике, из 100 тысяч болеет этим недугом 17 детей. Причиной является наследственное нарушения гена. Эту болезнь еще называют семейным гломерулонефритом.
 Cиндром Aльпорта — cложная наследственная патология, при которой к дисфункции почек добавляется нарушение слуха и зрения.
Cиндром Aльпорта — cложная наследственная патология, при которой к дисфункции почек добавляется нарушение слуха и зрения.Общие сведения
Болезнь Альпорта впервые описана в 1927 году ученым Британии Артуром Альпортом. Синдром встречается редко. Данные статистики показывают, что у 3 детей из 100 именно он является причиной пограничной почечной недостаточности у детей, и реже — у взрослых. Этот синдром — самый распространенный вид нефрита. Типы наследственности бывают разные, но наиболее частая Х-сцепленная доминантная форма. Она вызывает тяжелую почечную недостаточность у детей мужского пола. Проявляться начинает с первых лет жизни, проблемы со слухом и зрением развиваются позже. Потеря зрения и слуха предшествует развитию тяжелого нарушения работы почек, возникает в позднем детстве или в юношеские годы.
Вернуться к оглавлениюКлассификация
Заболевание Альпорта разделено на 2 квалификации из-за способа наследования патологии. Первая, генетическая, в свою очередь делится на 3 типа наследственного нефрита:
- Х-сцепленная доминантная -до 80% случаев;
- аутосомно-рецессивная — до 15% случаев;
- аутосомно-доминантная — до 5% случаев.
 Синдром Альпорта так же может развиться вследствие осложнений воспаления почек.
Синдром Альпорта так же может развиться вследствие осложнений воспаления почек.Вторая классификация, основная, указывает на 3 формы заболевания почек:
- Нефрит. Сопутствующие патологии — гематурия, проблемы со зрением и ухудшение слуха.
- Нефрит с гематурией без сопутствующих осложнений слуха и зрения.
- Доброкачественная семейная гематурия.
При наличии нефритов в первом и втором случаях основной классификации неизбежно наступает тяжелая почечная недостаточность. В третьем случае, при доброкачественном течении синдрома Альпорта, осложнения отсутствуют. Профилактика симптоматики болезни Альпорта и отсутствие инфекционных заболеваний способствует в дальнейшем полноценной жизненной деятельности.
Вернуться к оглавлениюКакие причины?
 Наследственные мутации в клетках почек провоцируют возникновение синдрома Альпорта.
Наследственные мутации в клетках почек провоцируют возникновение синдрома Альпорта.Наследственные мутации генов приводят к данной патологии. Нарушается функция биосинтеза коллагена четвертого типа. Коллаген — главная строительная основа для построения мембран в почках, ушах и глазницах. Функция мембран заключается в том, чтобы укреплять, поддерживать и разделять ткани. При недостаточном количестве или полном отсутствии синтеза строительного материала (коллагена) почечные мембраны не могут качественно отфильтровывать токсины и переработанные продукты из крови. В мочу попадает неотфильтрованный белок и эритроциты. Присутствие белка в крови называется протеинурия, эритроцитов — гематурия. Если синтез белка очень нарушен, это вызывает тяжелую почечную недостаточность, а в худшем случае — отказ почек. Прекращение работы почек приводит к летальному исходу.
Вернуться к оглавлениюПатогенез
Зарождение и развитие синдрома Альпорта, как правило, протекает изначально незаметно и обнаруживается случайным путем к 5 годам. Наследственный нефрит обычно характеризуется симптомами гломерулонефрита, иногда к нему добавляется нефротический синдром или симптоматика пиелонефрита. На начальных стадиях почки функционируют нормально. Отмечается незначительное присутствие белка и эритроцитов в крови, иногда повышенный уровень лейкоцитов. Примесь крови в моче появляется волнообразными приступами — от максимального до минимального уровня. При учащенном мочеиспускании развивается гипертензия или нефротический синдром. Иногда у пациентов возникает расширение чашечно-лоханочной системы, аминоацидурия.
 Потеря слуха и зрения от дисфункции почек чаще проявляется у мальчиков до 10 лет.
Потеря слуха и зрения от дисфункции почек чаще проявляется у мальчиков до 10 лет.Нарушения слуха, вплоть до глухоты, имеет неврогенные корни. Возникает чаще всего у детей до 10 лет, преимущественно мужского пола. Патология слуха часто бывает первым симптомом болезни и усугубляется со временем. Отдельные пациенты теряют слух при нормальном функционировании почек. Как будет протекать заболевание, и каков будет исход, во многом зависит от пола больного. Мужчины склонны к раннему развитию гипертензии и хронической почечной недостаточности. Летальный исход при отсутствии лечения наступает в период от 15 до 30 лет. У женского пола болезнь Альпорта протекает чаще всего скрыто, отмечается наличие гематурического синдрома. Сопутствуют болезни проблемы со слухом. Другие патологии проявляются как последствия синдрома Альпорта. Физические нагрузки и переутомление приводят к быстрому развитию заболевания.
Вернуться к оглавлениюСимптомы синдрома Альпорта
Врачи разделяют признаки синдрома Альпорта на 2 вида. Первый вид — это почечные проявления, которые характеризуются наличием в крови белков и эритроцитов. Наличие так называемого изолированного мочевого синдрома обнаруживается со временем. Видны они становятся только в возрасте 4−5 лет, а иногда явные проявления обнаруживаются в 8−9 лет. Но незаметные для невооруженного глаза капли крови присутствуют в моче постоянно — бессимптомная микрогематурия. Присутствие крови в моче — постоянный симптом, характеризующий синдром Альпорта. Часто перенесенные инфекции проявляют этот признак. После перенесенной простуды через 1−2 дня в моче видна кровь. Протеинурия возникает у мальчиков в подростковом возрасте, у девочек она либо минимальна, либо отсутствует вовсе.
 Синдрома Альпорта находится в «спящем» режиме около 9 лет, а после начинается ухудшение работы почек, слуха, зрения.
Синдрома Альпорта находится в «спящем» режиме около 9 лет, а после начинается ухудшение работы почек, слуха, зрения.Позже проявляются внепочечные признаки синдрома Альпорта — ослабление слуха, зрения, задержка в физиологическом развитии, лейомиоматоз (явление крайне редкое, разрастаются волокна мышц). Иногда заметны врожденные нарушения в физиологии — соединенные вместе или лишние пальцы, деформация ушей. Развиваясь, болезнь Альпорта провоцирует сопутствующее развитие почечной недостаточности, которая проявляется в желтом оттенке и сухости кожного покрова, часто ощущается сухость в ротовой полости, снижается частота и количество мочеиспусканий.
Вернуться к оглавлениюТечение болезни у детей
На первых этапах заболевания ребенок не чувствует дискомфорта. Единственным симптомом будет стойкость наличия крови в моче, сначала вовсе не заметная. Этот симптом присутствует во всех случаях, усиливаясь после перенесенных инфекционных болезней. Далее нарушается парциальное функционирование почек. Ребенок чувствует слабость, происходит интоксикация организма, часто снижается слух. Внешне ребенок становится бледным, быстро утомляется, часто возникают головные боли. Нарушение слуха развивается в разные периоды времени, в ранние годы его можно обнаружить только при помощи специальных приборов. Тугоухость возникает к 10 годам.
Зрение нарушается в 15−20% у больных детей. Характерны патологии хрусталика, катаракты. Часто развивается миопия. Постоянным симптомом является наличие возле желтого тела ярких белых или желтых вкраплений. Острота зрения снижается в 67% случаев. Наследственный нефрит у детей вызывает задержку в физическом развитии.
Вернуться к оглавлениюДиагностика
 Подтвердить диагноз синдрома Альпорта врач может на основе исследования анализов мочи, проверки почек, зрения, слуха.
Подтвердить диагноз синдрома Альпорта врач может на основе исследования анализов мочи, проверки почек, зрения, слуха.Для диагностики синдрома Альпорта проводятся лабораторные анализы, основной из них — проверка на присутствие крови и белка в моче. Также с помощью анализа мочи выявляют почечную недостаточность. Проводят биопсию почечных тканей на наличие ультраструктурных патологий. Если после проведения биопсии остаются сомнения, совершается генетический анализ, который выявляет мутации генов. Если возникло подозрение на наличие синдрома Альпорта, в обязательном порядке делают аудиометрию, что позволяет обнаружить нарушения слуха на самых ранних стадиях. Офтальмологические обследования фиксируют патологии зрения. Почечные патологии диагностируют с помощью ультразвукового обследования почек.
Вернуться к оглавлениюЛечение
Лечение синдрома проводится с появлением симптомов. При пиелонефрите используются антибактериальные и противовоспалительные медикаменты. Если развилась хроническая почечная недостаточность, применяется гемодиализ и необходима пересадка почек. На начальной стадии болезни без выраженных симптомов применяется профилактическое лечение:
 Блокируют дальнейшее развитие синдрома Альпорта диетотерапией, приёмом витаминов, очищением травами, исключением сильных нагрузок.
Блокируют дальнейшее развитие синдрома Альпорта диетотерапией, приёмом витаминов, очищением травами, исключением сильных нагрузок.- запрещена физическая активность, занятия спортом;
- частые прогулки на свежем воздухе положительно влияют на иммунитет;
- необходимо обеспечить полноценное питание с необходимым количеством витаминов и минералов;
- лечение травяным чаем — фитотерапия для снижения уровня эритроцитов в моче;
- прием витаминов А, Е и В6 — они улучшают обмен веществ.
Активно разрабатывается генная терапия синдрома Альпорта. В перспективе она может стать основным и наиболее эффективным лечением. Все методы лечения основаны на предупреждении и замедлении ухудшения функционирования почек. Диета должна содержать необходимое количество белка, жиров и углеводов. В случае нарушения функций почек нужно ограничить потребление фосфора и кальция. Необходимо остерегаться инфекционных заболеваний и избегать простуды. Инфекция может вызвать прогрессирование синдрома. Запрещается делать детям стандартные прививки. Профилактическое лечение проводится по 2−3 курса за год.
Вернуться к оглавлениюКакой прогноз?
Чаще всего неблагоприятный прогноз имеет наследственный нефрит с быстрым прогрессированием хронической почечной недостаточности. В зону риска попадают дети мужского пола с высоким уровнем крови и белка в моче. В этом случае наблюдается раннее развитие патологий зрения и слуха, функций почек, что требует лечения путем пересадки органов. Аутосомно-доминантная форма течения имеет благоприятный прогноз. Если поставлен диагноз изолированная гематурия без сопутствующих патологий слуха и при нормальном уровне белка в моче, хроническая почечная недостаточность не развивается и лечение должно быть направлено на предупреждение симптомов.
Синдром Альпорта у детей: лечение, диагностика — Симптомы
Причины синдрома Альпорта
Генетическая основа болезни — мутация в гене а-5 цепи коллагена IV типа. Этот тип универсален для базальных мембран почки, кохлеарного аппарата, капсулы хрусталика, сетчатки и роговицы глаза, что доказано в исследованиях с использованием моноклональных антител против этой фракции коллагена. В последнее время указывают на возможность применения ДНК-зондов для пренатальной диагностики наследственного нефрита.
Подчеркивается важность тестирования всех членов семьи с помощью ДНК-зондов для выявления носителей мутантного гена, что имеет большое значение при проведении медико-генетического консультирования семей с этим заболеванием. Однако до 20% семей не имеют родственников, страдающих болезнью почек, что позволяет предполагать высокую частоту спонтанных мутаций аномального гена. У большинства больных наследственным нефритом в семьях имеются лица с почечными заболеваниями, снижением слуха и патологией зрения; имеют значение родственные браки между людьми, имеющими одного или более предков, так как в браке родственных особей возрастает вероятность получения одинаковых генов со стороны обоих родителей. Установлены аутосомно-доминантный и аутосомно-рецессивный и доминантный, сцепленный с Х-хромосомой пути передачи.
У детей чаще различают три варианта наследственного нефрита: синдром Альпорта, наследственный нефрит без тугоухости и семейная доброкачественная гематурия.
Синдром Альпорта — наследственный нефрит с поражением слуха. В основе лежит сочетанный дефект структуры колагена базальной мембраны клубочков почек, структур уха и глаза. Ген классического синдрома Альпорта расположен в локусе 21-22 q длинного плеча Х-хромосомы. В большинстве случаев наследуется по доминантному типу, сцепленному с Х-хромосомой. В связи с этим у мужчин синдром Альпорта протекает тяжелее, так как у женщин функция мутантного гена компенсируется здоровым аллелем второй, неповрежденной хромосомы.
Генетической основой развития наследственного нефрита являются мутации в генах альфа-цепей коллагена IV типа. Известно шесть а-цепей IV коллагена Г типа: гены а5- и а6-цепей (Соl4A5 и Соl4A5) находятся на длинном плече Х-хромосомы в зоне 21-22q; гены а3- и а4-цепей (Соl4A3 и Соl4A4) — на 2-й хромосоме; гены a1- и a2-цепей (Соl4A1 и Соl4A2) — на 13-й хромосоме.
В большинстве случаев (80-85%) выявляется Х-сцепленный тип наследования заболевания, связанный с повреждениями гена Соl4A5 в результате делеции, точечных мутаций или нарушений сплайсинга. В настоящее время найдено более 200 мутаций гена Соl4A5, ответственных за нарушение синтеза а5-цепей коллагена IV типа. При этом типе наследования заболевание проявляется у детей обоего пола, но у мальчиков протекает тяжелее.
Мутации в локусах генов Соl4A3 и Соl4A4, ответственных за синтез а3- и а4 — цепей коллагена IV типа, наследуются аутосомно. По данным исследований аутосомно-доминантныи тип наследования отмечается в 16% случаев наследственного нефрита, аутосомно-рецессивный — у 6% больных. Изестно около 10 вариантов мутаций генов Соl4A3 и Соl4A4.
Результатом мутаций является нарушение процессов сборки коллагена IV типа, приводящее к нарушению его структуры. Коллаген IV типа является одним из основных компонентов гломерулярной базальной мембраны, кохлеарного аппарата и хрусталика глаза, патология которых будет выявляться в клинике наследственного нефрита.
Коллаген IV типа, входящий в состав гломерулярной базальной мембраны, состоит в основном из двух а1-цепей (IV) и одной а2-цепи (IV), а также содержит а3, а4, а5-цепи. Наиболее часто при Х-сцепленном наследовании мутация гена Соl4A5 сопровождается отсутствием а3-, а4-, а5- и а6 цепей в структуре коллагена IV типа, а количество о1- и а2-цепей в гломерулярной базальной мембране возрастает. Механизм этого феномена неясен, предполагается, что причиной являются посттранскрипционные изменения мРНК.
Отсутствие а3-, а4- и а5-цепей в структуре IV типа коллагена базальных мембран клубочков приводит к их истончению и ломкости на ранних стадиях синдрома Альпорта, что клинически проявляется чаще гематурией (реже гематурией с протеинурией или только протеинурией), снижением слуха и лентиконусом. Дальнейшее прогрессирование заболевания приводит к утолщению и нарушению проницаемости базальных мембран на поздних стадиях заболевания, с разрастанием в них коллагена V и VI типов, проявляющихся в нарастании протеинурии и снижении почечных функций.
Характер мутации, лежащей в основе наследственного нефрита, во многом определяет его фенотипическое проявление. При делеции Х-хромосомы с одновременной мутацией генов Соl4A5 и Соl4A6, ответственных за синтез а5- и а6-цепей коллагена IV типа, синдром Альпорта сочетается с лейомиоматозом пищевода и половых органов. По данным исследований при мутации гена Соl4A5, связанной с делецией, отмечаются большая тяжесть патологического процесса, сочетание почечного поражения с экстраренальными проявлениями и ранним развитием хронической почечной недостаточности, по сравнению сточечной мутацией этого гена.
Морфологически при электронной микроскопии выявляется истончение и расслоение гломерулярных базальных мембран (особенно lamina densa) и наличие электронно-плотных гранул. Поражение гломерул может быть неоднородным у одного и того же больного, от минимального фокального поражения мезангия до гломерулосклероза. Гломерулит при синдроме Альпорта всегда носит иммунонегативный характер, что отличает его от гломерулонефрита. Характерны развитие атрофии канальцев, лимфогистиоцитарная инфильтрация, наличие «пенистых клеток» с включениями липидов — липофагами. При прогрессировании заболевания выявляется утолщение и выраженная деструкция базальных мембран клубочков.
Выявляются определенные сдвиги в состоянии имунной системы. У больных наследственным нефритом отмечено снижение уровня Ig A и склонность к повышению концентрации IgМ в крови, уровень IgG может быть повышен на ранних стадиях развития заболевания и снижаться на поздних сроках. Возможно, повышение концентрации IgM и G является своеобразной компенсаторной реакцией в ответ на дефицит IgA.
Функциональная активность системы Т-лимфоцитов снижена; отмечается избирательное снижение В-лимфоцитов, ответственных за синтез Ig A, нарушается фагоцитарное звено иммунитета, в основном за счет нарушения процессов хемотаксиса и внутриклеточного переваривания в нейтрофилах
При исследовании биоптата почек у больных с синдромом Альпорта по данным электронной микроскопии наблюдаются ультраструктурные изменения базальной мембраны клубочка: истончение, нарушение структуры и расщепление гломерулярных базальных мембран с изменением ее толщины и неравномерностью контуров. На ранних стадиях наследственного нефрита дефект определяет истончение и ломкость гломерулярных базальных мембран.
Истончение гломерулярных мембран является более благоприятным признаком и чаще встречается у девочек. Более постоянный электронно-микроскопический признак при наследственном нефрите — расщепление базальной мембраны, причем выраженность деструкции ее коррелирует с тяжестью процесса.
Причины и механизм развития синдрома Альпорта
Синдром Альпорта вызван мутациями в генах COL4A4, COL4A3, COL4A5, отвечающих за биосинтез коллагена. Мутации в указанных генах нарушают нормальный синтез коллагена типа IV, который является очень важным структурным компонентом базальных мембран в почках, внутреннем ухе и глазах.
Базальные мембраны – это тонкие пленочные структуры, которые поддерживают ткани и отделяют их друг от друга. При нарушении синтеза коллагена типа IV гломерулярные базальные мембраны в почках не способны нормально фильтровать токсичные продукты из крови, пропуская в мочу белки (протеинурия) и эритроциты (гематурия). Аномалии синтеза коллагена типа IV приводят к почечной недостаточности и отказу почек, что и является главной причиной смерти при синдроме Альпорта.
Клиника
Гематурия – это наиболее частое и раннее проявление синдрома Альпорта. Микроскопическая гематурия наблюдается у 95% женщин и практически у всех мужчин. У мальчиков гематурию обычно выявляют в первые годы жизни. Если у мальчика за первые 10 лет жизни не обнаружена гематурия, то американские эксперты рекомендуют считать, что у него маловероятно наличие синдрома Альпорта.
Протеинурия в детстве обычно отсутствует, но иногда развивается у мальчиков с Х-сцепленным синдромом Альпорта. Протеинурия, как правило, прогрессирует. Значительная протеинурия у больных женского пола встречается нечасто.
Гипертензия чаще присутствует у пациентов мужского пола с XLAS и у больных обоих полов с ARAS. Частота и тяжесть гипертензии повышается с возрастом и по мере прогрессирования почечной недостаточности.
Нейросенсорная тугоухость (нарушение слуха) – это характерное проявление синдрома Альпорта, которое наблюдается довольно часто, но не всегда. Есть целые семьи с синдромом Альпорта, которые страдают от тяжелой нефропатии, но имеют нормальный слух. Нарушение слуха никогда не обнаруживается при рождении. Билатеральная высокочастотная нейросенсорная тугоухость обычно проявляется в первые годы жизни или в раннем подростковом возрасте. На ранней стадии болезни нарушение слуха определяется только при аудиометрии.
По мере прогрессирования, нарушение слуха распространяется на низкие частоты, включая человеческую речь. После появления тугоухости следует ожидать вовлечения почек. Американские ученые утверждают, что при Х-связанном синдроме Альпорта 50% мужчин страдают нейросенсорной тугоухостью к 25 годам, а к 40 годам – около 90%.
Передний лентиконус (выпячивание центрального участка хрусталика глаза вперед) наблюдается у 25% пациентов с XLAS. Лентиконуса нет при рождении, но с годами он приводит к прогрессирующему ухудшению зрения, которое заставляет больных часто менять очки. Состояние не сопровождается болью в глазах, покраснением или нарушениями цветового зрения.
Ретинопатия – это самое распространенное проявление синдрома Альпорта со стороны органа зрения, поражает 85% мужчин с Х-сцепленной формой болезни. Появление ретинопатии обычно предшествует почечной недостаточности.
Задняя полиморфная дистрофия роговицы – редкое состояние при синдроме Альпорта. У большинства нет никаких жалоб. Мутация L1649R в гене коллагена COL4A5 может также вызывать истончение сетчатки, которое ассоциируется с Х-сцепленным синдромом Альпорта.
Диффузный лейомиоматоз пищевода и бронхиального дерева – еще одно редкое состояние, которое наблюдается в некоторых семьях с синдромом Альпорта. Симптомы появляются в позднем детском возрасте и включают нарушение глотания (дисфагия), рвоту, боль в эпигастрии и за грудиной, частые бронхиты, одышку, кашель. Лейомиоматоз подтверждается компьютерной томографией или МРТ.
Аутосомно-рецессивная форма синдрома Альпорта
На ARAS приходится всего 10-15% случаев болезни. Эта форма встречается у детей, чьи родители являются носителями одного из пораженных генов, сочетание которых у ребенка вызывает болезнь. Сами родители не имеют симптомов или имеют незначительные проявления, а дети тяжело больны – их симптомы напоминают XLAS.
Аутосомно-доминантная форма синдрома Альпорта
ADAS – это самая редкая форма синдрома, которая затрагивает одно поколение за другим, причем мужчины и женщины болеют одинаково тяжело. Почечные проявления и глухота напоминают XLAS, но почечная недостаточность может возникать в более позднем возрасте. Клинические проявления ADAS дополняются склонностью к кровотечениям, макротромбоцитопенией, синдромом Эпштейна, наличием нейтрофильных включений в крови.
Диагностика синдрома Альпорта
• Лабораторные анализы. Анализ мочи: у больных с синдромом Альпорта чаще всего присутствует кровь в моче (гематурия), а также высокое содержание белка (протеинурия). Анализы крови демонстрирует почечную недостаточность.
• Биопсия тканей. Ткань почек, полученную при биопсии, исследуют с помощью электронной микроскопии на наличие ультраструктурных аномалий. Биопсия кожи менее инвазивна, и американские эксперты рекомендуют выполнять ее в первую очередь.
• Генетический анализ. В диагностике синдрома Альпорта, если остаются сомнения после биопсии почки, генетический анализ используется для получения однозначного ответа. Определяются мутации генов синтеза коллагена типа IV.
• Аудиометрия. Все дети с семейной историей, позволяющей заподозрить синдром Альпорта, должны проходить высокочастотную аудиометрию для подтверждения нейросенсорной тугоухости. Рекомендуется периодический мониторинг.
• Обследование глаз. Обследование у офтальмолога очень важно для раннего выявления и мониторинга переднего лентиконуса и других аномалий.
• УЗИ почек. На поздних стадиях синдрома Альпорта ультразвуковое исследование почек помогает выявить структурные нарушения.
Британские специалисты, основываясь на новых данных (2011) по генетическим мутациям у пациентов с Х-сцепленным синдромом Альпорта, рекомендуют анализ на мутации гена COL4A5, если пациент отвечает хотя бы двум диагностическим критериям по Gregory, и анализ COL4A3 и COL4A4, если мутация COL4A5 не обнаружена или подозревается аутосомное наследование.
Лечение синдрома Альпорта
Синдром Альпорта пока неизлечим. Исследования показали, что ингибиторы АПФ могут уменьшать протеинурию и замедлять прогрессирование почечной недостаточности. Таким образом, использование ИАПФ целесообразно у пациентов с протеинурией, независимо от наличия гипертензии. То же самое касается антагонистов АТII-рецепторов. Оба класса препаратов, судя по всему, помогают уменьшить протеинурию путем снижения внутриклубочкового давления. Более того, ингибирование ангиотензина-II, ростового фактора, отвечающего за гломерулярный склероз, теоретически может замедлять склерозирование.
Некоторые исследователи предполагают, что циклоспорин способен уменьшать протеинурию и стабилизировать почечную функцию у пациентов с синдромом Альпорта (исследования были небольшими). Но отчеты говорят, что ответ пациентов на циклоспорин очень вариабельный, и иногда препарат может ускорять интерстициальный фиброз.
При почечной недостаточности стандартная терапия включает эритропоэтин для лечения хронической анемии, препараты для контроля остеодистрофии, коррекцию ацидоза и антигипертензивную терапию для контроля АД. Применяется гемодиализ и перитонеальный диализ. Больным с синдромом Альпорта трансплантация почки не противопоказана: опыт пересадки в США показал хорошие результаты.
Генная терапия при разных формах синдрома Альпорта является перспективным вариантом лечения, который сегодня активно изучается западными медицинскими лабораториями.
Истинные причины заболевания
Истинные причины синдрома альпорта до сих полностью не изучены учеными. В нашем организме есть ген, функциональной обязанностью которого является обмен белка в тканях почек. Так вот мутация этого гена и является наиболее вероятной причиной появления недуга.
Теперь рассмотрим провоцирующие факторы, которые могут способствовать появлению заболевания. К ним можно отнести:
- тяжелые инфекционные процессы;
- прививки;
- сильные физические нагрузки.
Как видно из многочисленных случаев медицинской практики, порой развитию синдрома альпорта может способствовать обычная острая респираторная вирусная инфекция. Именно ввиду таких высоких рисков заболеваемости дети, которые имеют отягощенную наследственность, должны чаще проходить регулярное диагностическое обследование.
Впервые это заболевание было зарегистрировано в начале прошлого столетия. Врач наблюдал за семьей, в которой в течение нескольких поколений наблюдалась гематурия. Позже была замечена связь между гематурией и тугоухостью, а также поражением глаз. Позже, когда медицина совершенствовалась, медиками глубже исследовали генетическую природу данного синдрома.
В большинстве случаев у ее «обладателей» имеются родственники с почечными патологиями и другими признаками данного синдрома. Роль играют также родственные браки, в результате которых у ребёнка возрастает вероятность получения одинаковых генов. У больных с синдромом Альпорта выявляются сдвиги со стороны иммунной системы.
Симптомы
Наследственный нефрит имеет ярко выраженную клиническую симптоматику. Если говорить о начальных стадиях патологического процесса, то он проявляется следующим образом:
- появление крови в моче;
- ухудшение зрительной функции;
- нарушения слуховой способности, вплоть до развития глухоты.
Клинические симптомы будут нарастать по мере развития заболевания. С течением времени появляются признаки интоксикации, а также развивается анемия. Общее состояние и возраст пациента влияет на выраженность клинической картины.
Другими характерными симптомами заболевания являются такие признаки:
- сильные головные боли;
- мышечные боли;
- даже небольшая физическая активность быстро утомляет пациента;
- головокружение;
- артериальная гипертензия, которая сменяется резким падением давления;
- одышка;
- поверхностное дыхание;
- шум в ушах, который приобретает постоянный характер.
Если же речь идет о хронической форме наследственного нефрита, клиническая картина здесь будет немного отличаться, а именно:
- слабость и общее недомогание;
- частые позывы к мочеиспусканию, примеси крови;
- мочеиспускание не приносит облегчения;
- тошнота и рвота;
- ухудшение аппетита и, как следствие, потеря веса;
- кровоизлияние;
- зуд кожи;
- судороги;
- болезненные ощущения в грудной клетке;
- в тяжелых случаях появляется спутанность сознания и приступы беспамятства.
Инфекционные процессы дыхательных путей, активные физические нагрузки, профилактические прививки – все это может спровоцировать усиление гематурии. Что касается присутствия белка в моче, то сначала протеинурия имеет непостоянный характер, а затем постепенно нарастает и становится стойкой.
Нарастают также симптомы интоксикации, тугоухость особенно наблюдается у мальчиков, дети быстро устают, их беспокоят сильные головные боли. Дети значительно отстают в физическом развитии.
Виды
Специалисты различают три разновидности синдрома альпорта:
- ярко выраженные симптомы и быстрое прогрессирование острой почечной недостаточности;
- быстро прогрессирует заболевание, но при этом не наблюдается нарушение зрения и слуха;
- доброкачественное течение недуга, при котором отсутствуют клинические симптомы и прогрессирование. В этом варианте развития прогноз благоприятен. Если же у женщины присутствует аутосомно-рецессивный тип наследования, тогда наблюдается более тяжелое течение недуга.
Диагностическое обследование
Если присутствуют подозрения на наследственный фактор у детей, следует как можно раньше обратиться к детскому врачу. Для уточнения диагноза применяются лабораторные и инструментальные методы исследования. Что касается лабораторной диагностики, то она включает в себя общий анализ крови и мочи, а также биохимическое исследование.
Если говорить об инструментальной диагностике, то сюда можно отнести следующее:
- ультразвуковое исследование почек и надпочечников;
- биопсию почек;
- рентгенограмму почек.
Иногда могут понадобиться дополнительные генетические анализы. Больным назначается консультация нефролога и дополнительно – генетика.
Основными критериями диагностического обследования являются:
- присутствие в семье двух человек с нефропатией;
- гематурия является доминирующим симптомом;
- тугоухость у кого-то из членов семьи;
- появление хронической почечной недостаточности у кого-нибудь из родственников.
Если говорить о дифференциальном анализе, то наследственный нефрит сравнивают с приобретенной формой гломерулонефрита, при котором наблюдается также гематурия. В чем же отличие? Гломерулонефрит имеет острое начало и существует прямая связь с перенесенной инфекцией. Если наследственный нефрит проявляется в виде артериальной гипотонии, то гломерулонефрит, напротив, выражается в артериальной гипертензии.
Методы борьбы
Лечение синдрома альпорта включает в себя сочетание медикаментозных средств и специального диетического питания. Стоит отметить, что специфических лекарственных средств, которые бы устраняли именно этот генетический недуг, до сих пор не существует. Направленность лекарственных средств, применяемых при наследственном нефрите, связана с нормализацией работы почек. Диетическое питание для детей прописывается врачом индивидуально. Как правило, таких предписаний необходимо придерживаться всю оставшуюся жизнь. Показаны прогулки на свежем воздухе. В крайних случаях специалист может принять решение о проведении оперативного вмешательства. Обычно операция проводится при достижении пятнадцатилетнего возраста.
Правильное питание
Сразу же хочется отметить продукты питания, которые необходимо исключить из рациона. К ним относятся:
- соленая, жирная и копченая пища;
- пряности и острая еда;
- алкогольные напитки, но иногда врачи могут назначать красное вино в лечебных целях;
- продукты, которые имеют в своем составе искусственные красители.
Пища должна быть витаминизированной и калорийной, но при этом без высокого содержания белка. Физические нагрузки исключаются. Занятия спортом, особенно это касается детей, могут проходить только в том случае, если они не запрещены врачом.
Пища должна быть полноценной и в достаточном количестве содержать белки, жиры и углеводы, при этом должны учитываться функциональные способности почек.
Почечная недостаточность является одним из наиболее тяжелых осложнений синдрома альпорта. Согласно статистике, недостаточностью страдают мальчики от шестнадцати до двадцати лет. Если будет отсутствовать адекватное лечение и правильный способ жизни, то летальный исход наступает раньше, чем в тридцать лет.
Кроме этого, важно избегать контакта с инфекционными больными для снижения рисков развития респираторных заболеваний. Профилактические прививки противопоказаны детям с наследственным нефритом, а вакцинация может проводиться лишь по эпидемиологическим показаниям.
Профилактики синдрома альпорта нет. Это генетический недуг, который предупредить невозможно. Если же у ребенка был диагностирован недуг, тогда следует придерживаться рекомендаций врача и правильного образа жизни.
Если говорить о прогнозах, то крайне неблагоприятными являются следующие критерии:
- мужской пол;
- наличие хронической почечной недостаточности у членов семьи;
- неврит слухового нерва;
- наличие белка в моче.
Больным назначаются лекарственные средства, которые влияют на улучшение обмена веществ:
- витамин А, Е;
- пиридоксин;
- АТФ;
- кокарбоксилаза.
Трансплантация – это наиболее эффективный метод лечения при синдроме альпорта. Рецидив заболевания в трансплантате не отмечается, и лишь в незначительных случаях возможно развитие нефрита.
Итак, синдром альпорта – это серьезный недуг, требующий своевременного и грамотного подхода к лечению. Не существует профилактики наследственного нефрита, но его течение можно облегчить при неукоснительном соблюдении всех врачебных рекомендаций.
Синдром Альпорта у детей: что собой представляет, причины, симптомы, методы диагностики и лечения, а также прогноз и последствия, фото
Из-за активно развития различных отраслей промышленности атмосфера, вода и почва регулярно загрязняются химическими веществами, а также подвергаются воздействию облучения. Это увеличивает количество возможных генетических мутаций и наследственных аномалий, которые появляются у пациентов в совсем юном возрасте. К одним из таких недугов относят синдром Альпорта, при котором поражается преимущественно мочевыделительная система.
Определение синдрома Альпорта
Синдром Альпорта — это наследственное заболевание, которое сопровождается генетическими мутациями. Клинические проявления патологического состояния крайне разнообразны: у пациентов встречаются воспалительные изменения почечных канальцев (90% случаев), нарушения слуха (60%), зрения (20%) и внешние уродства. Чаще всего недуг диагностируется в юном возрасте на фоне частых обращений к доктору по поводу проблем с мочевыделительной системой.
Заболевание поражает преимущественно почки
Синдром Альпорта относится к разряду довольно редких патологий и встречается у 20 из 100 тысяч детей, мальчики страдают чаще девочек.
Взрослые люди без каких-либо симптоматических проявлений недуга могут являться носителями мутантного гена. При его передаче потомству патология возникает в любом из последующих поколений. Вероятность рождения ребёнка с такой мутацией увеличивается при близкородственных браках.
Причины возникновения болезни
Синдром Альпорта — это следствие генетической мутации. В результате её развития в организме начинает синтезироваться аномальный коллаген, который входит в состав почечных мембран и канальцев, органов слуха и зрения, что обуславливает появление клинической симптоматики. Существуют 3 типа наследования заболевания:
- аутосомно-доминантный (если страдает хотя бы один из родителей, недуг 100% будет у ребёнка),
- аутосомно-рецессивный (для рождения больного малыша необходим союз двух людей, являющихся носителями мутантного гена),
- сцепленный с половой хромосомой (передаётся от матери плоду преимущественного мужского пола, так как девушки имеют генотип XX, а мужчины — XY).
Какие факторы, воздействующие на беременную женщину, увеличивают вероятность развития недуга:
- облучение (рентгеновское, ультрафиолетовое),
- радиация,
- проживание в экологически неблагоприятных условиях (загрязнение воды, почвы, атмосферы),
- самовольный приём фармацевтических препаратов (в особенности антибиотиков, гормонов, цитостатиков, иммунодепрессантов),
- работа на вредном производстве (контакт с ядовитыми газами, тяжёлыми металлами),
- употребление наркотиков,
- алкоголизм и курение.
Основные симптомы патологии
Заболевание развивается довольно остро, в результате чего обнаружить его можно в первые 2–4 года жизни малыша. При рождении у детей могут быть определённые внешние уродства (заячья губа, волчья пасть, изменение размеров черепа), которые наталкивают докторов на этот диагноз. К основным клиническим проявлениям синдрома относятся:
- боль в поясничной области, усиливающаяся при ходьбе, беге, интенсивных физических нагрузках,
- появление в урине крови, мутности и патологического осадка из-за белков и лейкоцитов,
- рези при мочеиспускании,
- прогрессирующее снижение слуха или полное его отсутствие при рождении,
- ухудшение зрения (близорукость),
- повышение артериального давления,
- отёки мягких тканей (преимущественно в утреннее время суток).
Далеко не всегда синдром Альпорта сопровождается всеми симптомами. К одному из моих знакомых докторов пришли родители 7-летнего мальчика, у которого наблюдалось прогрессирующее снижение остроты зрения. Офтальмологи не смогли найти причину и отправили малыша на дополнительное обследование. После пройденных анализов было выявлено наследственное заболевание, а затем медики начали необходимое лечение.
Фотогалерея: дети с синдромом Альпорта

Изменение формы лицевого черепа часто встречается при синдроме Альпорта
Одутловатость лица — следствие задержки воды в тканях
Снижение слуха возникает у большинства пациентов
Методы диагностики
Родители маленьких пациентов часто обращаются с первыми симптомами патологии к нефрологу, педиатру, ЛОР-врачу или офтальмологу. Доктора проводят осмотр, выявляя внешние дефекты, а также изучают данные семейного анамнеза.
Если один из бабушек, дедушек, братьев, сестёр или родителей ребёнка имел подобную патологию, вероятность развития её у малыша составляет 80%.
Не забывайте, что приносить мочу на анализ можно только в специальной стерильной ёмкости. Во время работы в лаборатории мне часто приходилось сталкиваться с тем, что пациенты помещали отходы жизнедеятельности ребёнка в баночки из-под пищевых продуктов. Даже при тщательной обработке с моющими средствами в них остаются частицы белков и жиров, которые существенно затрудняют обработку биологического материала. Подобные загрязнения дают недостоверный результат, приходится повторно направлять пациентов на обследование.
Каким образом подтверждают синдром Альпорта:
Таблица: круг заболеваний для дифференциальной диагностики недуга
Различные варианты лечения патологии
Терапия синдрома Альпорта проводится пожизненно. Пациенты с лёгкой стадией развития недуга и незначительным поражением слухового анализатора могут получать фармацевтические препараты в домашних условиях и регулярно госпитализироваться в стационар для дообследования и контроля. У больных с выраженными нарушениями функции почек могут понадобиться более радикальные меры, включающие гемодиализ и трансплантацию. Для коррекции нарушений слуха можно использовать специальные аппараты или кохлеарные импланты.
Основные цели лечения детей с синдромом Альпорта:
- обеспечение нормального физического и психоэмоционального развития,
- интеграция в общество и коллективы,
- нормализация функции почек,
- профилактика вторичных осложнений,
- стабилизация уровня артериального давления.
Видео: доктора рассказывают о борьбе с почечной недостаточностью
Общие рекомендации по режиму
Пациентом с синдромом Альпорта показаны некоторые ограничения в обычной жизнедеятельности. Это поможет оградить малыша от преждевременного развития обострений и способствует полноценной адаптации в обществе.
Правила предосторожности:
- не записывайте ребёнка в травмоопасные секции (к ним относятся хоккей, футбол, волейбол, баскетбол, борьба, гимнастика),
- с раннего возраста приучайте к правильному питанию и контролю за количеством выпитой жидкости,
- регулярно проходите осмотр у ЛОра и офтальмолога, чтобы корректировать возникающие нарушения органов чувств,
- не ограничивайте контакт малыша со сверстниками: он должен расти и развиваться соответственно возрасту,
- при наличии отставания в психофизическом плане от других детей обратитесь за помощью к педиатру.
Таблица: медикаментозные препараты для борьбы с недугом
Фотогалерея: препараты для лечения синдрома Альпорта

Ибуклин снимает воспаление и отёк

Реополиглюкин нормализует солевой баланс

Пиридоксин восстанавливает потребность организма в витамине В6
Фитотерапия при заболевании
Чтобы вывести из организма малыша ненужные токсины и шлаки, которые накапливаются в течение жизни, необязательно всегда прибегать к использованию медикаментов. С возраста 2–3 лет допускается применение природных средств, приготовленных на основе трав, растений и плодов. Они также эффективно справляются с воспалительным процессом и способствуют укреплению детского организма.
Перед началом употребления любого настоя или отвара проведите аллергическую пробу. Для этого достаточно нанести несколько капель на сгиб запястья малыша. При отсутствии реакции разрешено продолжать применение.
Самые популярные рецепты для борьбы с симптомами недуга:
- Несколько крупных морковок измельчите в соковыжималке. Удалите переработанную мякоть, добавьте немного сахара или мёда, а затем предложите такой напиток малышу после еды. Морковь обладает антиоксидантными и иммуностимулирующими свойствами, а также является источником витамина А, который улучшает зрение. Употреблять такой сок стоит 1 раз в неделю.
- 100 грамм шиповника поместите в кастрюлю с 2 литрами кипятка. Варите на протяжении 20 минут, постоянно помешивая. После остывания утром и вечером давайте ребёнку по 1 стакану напитка. Шиповник способен снять воспаление и избавить малыша от неприятных ощущений при мочеиспускании. Использовать этот способ нужно ежедневно.
- 1 пакетик аптечной ромашки заварите в стакане кипятка и дайте выпить малышу перед сном. Это средство обладает седативным и спазмолитическим эффектом, благодаря чему ребёнок будет чувствовать себя лучше. Можно применять такое средство ежедневно.
Фотогалерея: природная терапия недуга

Морковь содержит витамин А

Шиповник снимает воспаление

Ромашка обладает успокаивающим воздействием на нервную систему
Видео: натуральные средства для лечения нефрита
Правила питания для больных с синдромом Альпорта
Чтобы избежать излишней нагрузки на почки, маленьким пациентам необходимо придерживаться строгой диеты. Она должна быть сбалансированной и содержать белки, жиры, углеводы и витаминно-минеральные компоненты соответственно весу и возрастной группе ребёнка. Все блюда необходимо готовить на пару, тушить или запекать с минимальным количеством масла и поваренной соли. Продукты должны быть максимально свежими и натуральными, без вредных химических добавок.
Обязательно следите за количеством употребляемой ребёнком жидкости. Это позволит вовремя заподозрить патологические внутренние отёки и принять меры.
Какие продукты нужно исключить из рациона:
- майонез и все соусы на его основе,
- жирное мясо и масляную рыбу,
- консервы,
- чипсы и сухарики,
- фастфуд и полуфабриканты,
- газированные напитки и сладкие соки,
- глазированные сырки,
- колбасу, сосиски, сардельки.
Фотогалерея: вредная еда
Чипсы содержат много соли

Газированные напитки замедляют обмен веществ

Фастфуд содержит вредные жиры
Что необходимо добавить в питание:
- свежие овощи, фрукты и ягоды,
- орехи и мёд,
- молоко и молочные продукты,
- зелень,
- крупы (гречку, рис, овсянку, пшено),
- гороховые и бобовые,
- нежирное мясо и рыбу.
Фотогалерея: полезная еда

Молочные продукты — источник кальция

Крупы содержат полезные углеводы

Зелень богата витаминами
Показания для гемодиализа и трансплантации
При неблагоприятном течении болезни у пациента развивается недостаточность почек. Для увеличения продолжительности жизни таким детям необходимо проведение гемодиализа — процедуры по искусственному очищению крови от вредных примесей. Показаниями к такому лечению служат:
- неуклонное прогрессирование болезни,
- увеличение количества белка, выделяемого почками,
- угрожающее жизни состояние,
- повышение уровня токсинов в организме.
Гемодиализ проводится даже младенцам при угрожающем жизни состоянии
Трансплантация донорской почки доступна пациентам в возрасте от 6 лет (возможны исключения). Сущность этой операции заключается в пересадке донорского органа в тело больного ребёнка, что позволяет улучшить качество его жизни. Новая почка может функционировать в течение 15–19 лет, после чего необходима замена. Показаниями к этой операции служат:
- хроническая почечная недостаточность,
- наличие только 1 функционирующего органа,
- прогрессирующий воспалительный процесс.
Прогнозы лечения и возможные осложнения
Так как наследственные заболевания связаны с генетическим дефектом, полностью избавиться от его проявлений нереально. Ожидаемая продолжительность жизни пациента зависит от тяжести течения недуга. Известно, что больные с нарушениями слуха и внешними уродствами живут на 5–10 лет меньше. Неблагоприятными прогностическими факторами также служат:
- принадлежность к мужскому полу,
- частые обострения (6 и более раз в течение 1 года),
- обнаружение большого уровня белка в моче (2–3 грамма в литре).
Какие осложнения могут возникнуть у детей с синдромом Альпорта:
- Острая или хроническая недостаточность почек. Эта патология напрямую связана с нарушением способности органов мочевыделительной системы выполнять функцию фильтрации и реабсорбции полезных веществ. В результате в крови накапливается большое количество токсинов, которые повреждают головной мозг и сердце. Для устранения недуга применяются трансплантация почек и гемодиализ.
- Проблемы с психомоторным развитием. При синдроме Альпорта страдает слуховой анализатор, что может проявляться как тугоухостью, так и полной глухотой. Далеко не всегда удаётся обнаружить эту проблему на ранних стадиях, в результате чего малыш неполноценно контактирует с окружающим миром и хуже усваивает информацию. С целью профилактики этого осложнения используют слуховые аппараты и специальные корректирующие школы.
- Присоединение вторичной инфекции мочевыводящих путей. На фоне хронического воспаления почечных канальцев нередко возникают различные абсцессы, флегмоны и карбункулы, поражающие ткань органа. Лечение проводится хирургическим путём.
- Мочекаменная болезнь — образование в различных участках выделительной системы плотных конгломератов, затрудняющих ток мочи. Для профилактики этого осложнения детям с самого раннего возраста необходимо соблюдать диету.
- Гидронефроз — расширение почечной лоханки из-за скопления в ней большого количества жидкости. Возникает на фоне нарушения оттока урины и механического сдавления органа отёком.
- Увеличение риска развития злокачественных новообразований. Лица, страдающие от генетических аномалий, гораздо чаще подвергаются возникновению опухолей в области почек. Для устранения этого осложнения используют химиотерапию и хирургическое вмешательство.
Фотогалерея: нежелательные последствия недуга
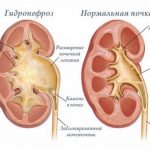
Гидронефроз — патологическое расширение лоханки из-за её растяжения
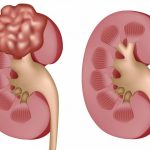
Опухоли почек у больных синдромом Альпорта носят злокачественный характер
Мочекаменная болезнь — это образование камней в почках, лоханках и мочеточниках
Способы профилактики осложнений
Генетические заболевания относятся к разряду тех патологий, предупредить развитие которых полностью невозможно. Ещё до зачатия ребёнка будущая мать должна ответственно подходить к вопросам своего здоровья, чтобы суметь своевременно предотвратить рождение больного малыша. Беременной женщине необходимо отказаться от употребления алкоголя, наркотических средств и курения, при наличии возможности — переехать в районы с благоприятной экологической ситуацией и уволиться с опасной работы. Вторичная профилактика синдрома Альпорта направлена на защиту от вредных воздействий окружающей среды и поддержание иммунитета.
В своей практике мне доводилось сталкиваться с тем, что некоторые родители перед заведением потомства проходили генетическое исследование. Это позволяло выявить у них наличие генов, которые отвечали за развитие синдрома Альпорта и других наследственных заболеваний, даже если сами будущие родители не имели никаких серьёзных проблем со здоровьем. У одной пары таким образом была обнаружена подобная мутация, а рассчитанный доктором показатель риска появления на свет больного малыша был крайне велик. Мужчина и женщина приняли решение об усыновлении ребёнка из детского дома.
Правила вторичной профилактики развития осложнений при синдроме Альпорта:
- защищайте малыша от различных переохлаждений и резких перепадов температуры,
- при отсутствии медицинских отводов выполняйте ежегодную вакцинацию (в особенности от вируса гриппа), чтобы избежать осложнений на почки,
- каждые полгода даже при отсутствии обострений сдавайте анализы мочи и крови: это позволит сразу же заподозрить ухудшение общего состояния,
- старайтесь, чтобы ребёнок больше времени проводил на свежем воздухе (на здоровье мочевыделительной системы хорошо влияет отдых на море или бальнеологических курортах),
- питайтесь только здоровой и правильной едой, отказавшись от фастфуда и вредных напитков,
- занимайтесь с малышом лечебной гимнастикой или любыми лёгкими спортивными играми,
- при необходимости приобретите слуховой аппарат или кохлеарный имплант,
- избегайте контакта с заболевшими простудой людьми,
- постоянно обследуйтесь у педиатра и нефролога.
Синдром Альпорта — это тяжёлая генетическая аномалия, которая приносит пациенту массу неудобств. Без получения ежедневной дозы фармацевтических препаратов значительно снижаются качество и продолжительность жизни. Современная медицина способна полностью адаптировать больного с синдромом Альпорта к обитанию в социуме, а также избавить от нежелательных осложнений недуга. Однако нужно помнить, что даже при стабилизации состояния и устойчивой ремиссии необходимо постоянно придерживаться правил профилактики и следить за своим здоровьем.
 Загрузка…
Загрузка…Другие уточненные синдромы врожденных аномалий, не классифицированные в других рубриках > Архив — Клинические протоколы МЗ РК
Тактика лечения: специфического лечения нет, симптоматическое лечение.
Цели лечения:
— активизация психического развития;
— пополнение пассивного и активного словарного запаса;
— коррекция поведения;
— повышение эмоционального тонуса, настроения ребенка;
— обучение навыкам самообслуживания;
— социальная адаптация.
Немедикаментозное лечение:
— индивидуальные занятия с логопедом;
— занятия с психологом;
— кондуктивная педагогика;
— малокалорийная диета;
— ЛФК;
— физиолечение.
Медикаментозное лечение
Широко используют в последнее время препараты ноотропного ряда — нейропротекторы, с целью улучшения обменных процессов в головном мозгу. Большинство ноотропных препаратов в связи с их психостимулирующим действием назначают в первую половину дня. Продолжительность курсов лечения ноотропами составляет от одного до двух-трех месяцев, церебролизин, ампулы 1 мл в/м, пирацетам, ампулы 5 мл 20%, гинкго-билоба (танакан), таблетки 40 мг, пиритинол гидрохлорид (энцефабол), драже 100 мг, суспензия — 5 мл содержат 80,5 мг пиритинола (соотв.100 мг пиритинола гидрохлорида).
Энцефабол — минимум противопоказаний, разрешен к применению с первого года жизни. Дозирование суспензии (с содержанием в 1 мл 20 мг энцефабола) детям 3-5 лет суточная доза 200-300 мг (12-15 мг массы тела) назначают в 2 приема — утром (после завтрака) и днем (после дневного сна и полдника). Продолжительность курса 6-12 недель (целесообразен длительный прием, при котором повышается работоспособность и способность к обучению, улучшаются высшие психические функции): актовегин, ампулы 2 мл 80 мг, драже-форте 200 мг активного вещества.
Нейрометаболический препарат, содержащий исключительно физиологические компоненты. Детям назначается в драже-форте, прием до еды по ½ -1 драже 2-3 раза в день (в зависимости от возраста и выраженности симптомов заболевания) до 17 часов. Продолжительность терапии 1-2 месяца. Инстенон, таблетки (1 таблетка содержит 50 мг этамивана, 20 мг гексобендина, 60 мг этофиллина). Многокомпонентный нейрометаболический препарат. Суточная доза составляет 1,5-2 таблетки, назначается в 2 приема (утром и днем) после еды. Для исключения побочных эффектов рекомендуется постепенное наращивание дозы в течение 5-8 дней. Продолжительность лечения 4-6 недель.
Ангиопротекторы с целью улучшения мозгового кровообращения: винпоцетин, циннаризин.
Седативная терапия по показаниям: ноофен, ново-пассит.
Корректоры поведения: сонапакс, хлорпротиксен.
Витамины группы В: В1, В6, В12, нейромультивит, неуробекс, фолиевая кислота, аевит.
Профилактические мероприятия:
— профилактика травматизма;
— профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение: регулярные занятия с логопедом, дефектологом, психологом, социальная адаптация ребенка, оформление в специализированный детский сад, прохождение медико-педагогической комиссии для решения вопроса об обучении ребенка.
Основные медикаменты:
1. Актовегин, ампулы 2 мл 80 мг
2. Винпоцетин, таблетки 5 мг
3. Пирацетам, ампулы 5 мл 20%
4. Пирацетам, таблетки 0,2 и 0,4
5. Пиридоксин гидрохлорид, ампулы, 1 мл 5%
6. Танакан, таблетки 40 мг
7. Тиамин хлорид, ампулы 5% 1 мл
8. Фолиевая кислота, таблетки 0,001
9. Церебролизин, ампулы 1 мл
10. Цианокобаламин, ампулы 1 мл 200 мкг и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Глицин, таблетки 0,1
3. Гопантеновая кислота (пантокальцин), таблетки 0,25
4. Инстенон, таблетки
5. Нейромультивит, таблетки
6. Неуробекс, таблетки
7. Ноофен,таблетки 0,25
8. Пиритинол, драже 0,1, суспензия
9. Сонапакс, таблетки 10 мг
10. Хлорпритиксен 15 мг
11. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
— улучшение внимания, памяти, работоспособности;
— пополнение пассивного и активного запаса слов;
— повышение эмоционального и психического тонуса.