ЖИРОВОЙ ГЕПАТОЗ ПЕЧЕНИ И ГЕПАТОПРОТЕКТОРЫ
ЖИРОВОЙ ГЕПАТОЗ ПЕЧЕНИ И ГЕПАТОПРОТЕКТОРЫ
Что такое жировой гепатоз и почему он развивается?
В нашем веке с развитием медицины появилось множество новых и малознакомых обывателю диагнозов, один из таких – жировой гепатоз. По данным врачей значительная часть человечества страдают в той или иной степени этим недугом. Такой диагноз подразумевает под собой, факт накопления печеночными клетками избыточного жира. Жировой гепатоз сопровождается нарушениями обмена питательных веществ в печеночной ткани (дистрофией). Наиболее распространённой причиной жировой дистрофии печени является избыточное употребление жиров и углеводов в пищу. Возможен и другой вариант: жиры могут избыточно накапливаться в крови вследствие различных нарушений обмена веществ, а также вследствие воздействия токсических веществ на печень – лекарств, алкоголя и др.
Итак, гепатоз — это нарушение обмена веществ в печёночных клетках, приводящее к развитию в клетках печени дистрофических изменений. Гепатоз может развиваться не только вследствие накопления жира, но и по причине избыточного поступления в печеночную клетку других веществ (например, пигментов). Пигментный гепатоз обусловлен нарушениями обмена билирубина и жёлчных кислот в печени. Пигментные гепатозы носят наследственный характер, проявляющийся хронической или периодически появляющейся желтухой. Изменения структуры тканей и функции печени не выражены.
Гепатоз может развиваться не только вследствие накопления жира, но и по причине избыточного поступления в печеночную клетку других веществ (например, пигментов). Пигментный гепатоз обусловлен нарушениями обмена билирубина и жёлчных кислот в печени. Пигментные гепатозы носят наследственный характер, проявляющийся хронической или периодически появляющейся желтухой. Изменения структуры тканей и функции печени не выражены.
Но именно жировой гепатоз — наиболее часто встречаемый вид гепатоза. Жировой гепатоз также называют жировой дистрофией печени, так как он сопровождается «ожирением» печёночных клеток. Крайне распространенная причина жирового гепатоза — токсическое воздействие на печёночные клетки алкоголем. Что происходит в этом случае? Дело в том, что этиловый спирт провоцирует превращение жирных кислот в жиры и приводит к накоплению их в печени.
Кроме того, обменные нарушения жира развиваются при эндокринных заболеваниях (например, заболевания, сопровождающиеся снижением количества веществ, вовлечённых в переработку жиров).
Жировая дистрофия печени может развиваться и вследствие нарушения выведения жира из печени.
Наиболее часто жировой гепатоз развивается совместно с сахарным диабетом, при различных формах нарушений белкового обмена, при ожирении и так называемом метаболическом синдроме (патологический комплекс, состоящий из ожирения, снижения чувствительности тканей к инсулину или его повышенное содержание, нарушения углеводного, липидного, пуринового обменов и артериальной гипертензии).
Симптомы жирового гепатоза и диагностика.
Гепатоз печени – это процесс, длящийся годами. Развивается он постепенно, при этом неуклонно нарастает недостаточность печёночных клеток. Появляются симптомы гепатоза печени спустя десятилетия и трудно идентифицируются с точки зрения диагностики, так имеют самый общий (неспецифический) характер: усталость, потеря веса и аппетита. Появляются и проявления нарушений со стороны желудочно–кишечного тракта: тошнота, рвота, метеоризм.
Если врач назначает лабораторные и инструментальные обследования, то обнаруживаться признаки воспаления печени. Изменения ее структуры. Проявляется это повышением уровня ферментов крови, свидетельствующих о разрушении клеток (повышаются ферменты печени: АСТ, АЛТ, ЛДГ и ЩФ). Эти ферменты являются составной частью клеток, поэтому, если клетки печени разрушаются, повышаются их значения в крови. Щелочная фосфатаза образуется в жёлчном пузыре и его протоках. Но для подтверждения диагноза гепатоза этого недостаточно. Как правило проводятся более детальные обследования, которые помогают установить факт дистрофических изменений. Поэтому доктор назначает комплекс исследований. При проведении УЗИ обнаруживается увеличение печени. Если гепатоз уже развился – обнаруживается «зернистость» паренхиматозной ткани.
Изменения ее структуры. Проявляется это повышением уровня ферментов крови, свидетельствующих о разрушении клеток (повышаются ферменты печени: АСТ, АЛТ, ЛДГ и ЩФ). Эти ферменты являются составной частью клеток, поэтому, если клетки печени разрушаются, повышаются их значения в крови. Щелочная фосфатаза образуется в жёлчном пузыре и его протоках. Но для подтверждения диагноза гепатоза этого недостаточно. Как правило проводятся более детальные обследования, которые помогают установить факт дистрофических изменений. Поэтому доктор назначает комплекс исследований. При проведении УЗИ обнаруживается увеличение печени. Если гепатоз уже развился – обнаруживается «зернистость» паренхиматозной ткани.
В сложных диагностических ситуациях проводят даже биопсию печени (берут небольшой кусочек печени и изучают его).
Гепатоз — как лечить?
Как и любой хронический процесс гепатоз лечится долго с применением комплексной терапии. Самое важное – устранить причину нарушения обмена жира. Если это токсическое вещество – исключить его прием, если неверный режим жизни — начать правильно питаться, вести нормальный образ жизни, взять под контроль уровень сахара крови и т.д. Но что делать с уже поврежденными клетками печени? Вот тут есть хорошая новость – печень способна восстанавливать свои клетки. Важно поддержать этот процесс. Дело в том, что в основе механизмов повреждения гепатоцита лежит активация перекисных процессов мембраны клетки. Поэтому, помимо диеты и лечения сопутствующей или первичной болезни, вызвавшей жировой гепатоз, необходимо провести восстановление гепатоцита и его мембраны. Для этого активно используют две группы препаратов:
Если это токсическое вещество – исключить его прием, если неверный режим жизни — начать правильно питаться, вести нормальный образ жизни, взять под контроль уровень сахара крови и т.д. Но что делать с уже поврежденными клетками печени? Вот тут есть хорошая новость – печень способна восстанавливать свои клетки. Важно поддержать этот процесс. Дело в том, что в основе механизмов повреждения гепатоцита лежит активация перекисных процессов мембраны клетки. Поэтому, помимо диеты и лечения сопутствующей или первичной болезни, вызвавшей жировой гепатоз, необходимо провести восстановление гепатоцита и его мембраны. Для этого активно используют две группы препаратов:
- гепатопротекторы — повышают устойчивость гепатоцитов к влиянию патологических факторов. Гепатопротекторы стабилизируют поврежденные мембраны клеток и другие важные органеллы.
- антиоксиданты — замедляют биохимические реакции окисления, в которых участвуют свободные радикалы. Свободные радикалы повреждают клеточные мембраны.
 Антиоксиданты нейтрализуют свободные радикалы и сохраняют мембрану клеток. Именно мембрана клетки подвержена перекисному окислению липидов, которых в ней множество.
Антиоксиданты нейтрализуют свободные радикалы и сохраняют мембрану клеток. Именно мембрана клетки подвержена перекисному окислению липидов, которых в ней множество.
 Антиоксиданты нейтрализуют свободные радикалы и сохраняют мембрану клеток. Именно мембрана клетки подвержена перекисному окислению липидов, которых в ней множество.
Антиоксиданты нейтрализуют свободные радикалы и сохраняют мембрану клеток. Именно мембрана клетки подвержена перекисному окислению липидов, которых в ней множество.Гепатопротекторов и антиоксидантов в свободной продажи очень много, выбрать подходящие комбинации поможет врач. Появляются и новые, обладающие комплексными эффектами, гепатопротекторы. Российскими учеными был разработан препарат гепатопротекторного ряда на основе полипренолов – натуральных веществ полученных высокотехнологичным способом из хвойных растений. Именно полипренолы обладают способностью оказывать антиоксидантное действие, не только защищая мембрану клеток, но и восстановляя нормальные процессы клеточного дыхания в энергетических станциях клеток – митохондриях. Очень важно то, что гепатопротектор на основе полипренолов показал способность снижать уровень липопротеинов очень низкой плотности, приводящих к атеросклерозу. В клинических исследованиях были показаны выраженные противовоспалительные эффекты при различных вариантах гепатоза. Быстрое и стойкое снижение маркеров цитолиза, о которых говорилось выше, свидетельствовали об этом.
Быстрое и стойкое снижение маркеров цитолиза, о которых говорилось выше, свидетельствовали об этом.
Лечение гепатоза длительный и кропотливый процесс, требующий грамотного подхода и тщательного выбора препаратов для восстановления печени. Поэтому приняв решение лечиться важно вникнуть во все детали заболевания, проконсультироваться с доктором и тогда приступать к терапии гепатоза.
Гепатозы пигментный — Справочник по медицине PRO7
Определение. Гипербилирубинемия Жильбера (ферментопатическая гипербилирубинемия, доброкачественная семейная негемолитическая гипербилирубинемия, наследственный пигментный гепатоз). [Стр.422]Этиология. Заболевание имеет врожденный характер и наследуется по доминантному типу. [Стр.422]
Пример формулировки диагноза. Синдром Жильбера. Гепатобио-псия от 3.05.02. [Стр.423]
Боль или чувство тяжести в правом подреберье наблюдаются часто, особенно в периоды обострений. [Стр.433]
Синдром Жильбера — наследственная гипербилирубинемия (пигментный гепатоз).
 [Стр.2]
[Стр.2]Наследственные пигментные гепатозы (болезни Жильбера, Дабина— Джонса, Ротора, Криглера—Найяра I и II типа). [Стр.777]
Дифференцируют ХГ и с наследственными пигментными гепатозами и обменными заболеваниями печени (см. ниже). [Стр.797]
Дифференциально-диагностические признаки различных форм пигментных гепатозов приведены в табл. 7. [Стр.135]
Морфологическая характеристика. При всех формах пигментных гепатозов печень сохраняет гистологическое строение, близкое к норме. Каких-либо признаков диспротеиноза, некроза в печеночных клетках, как правило, нет. [Стр.127]
Выделяют несколько патогенетически обусловленных типов пигментных гепатозов (функциональных гипербилирубинемий). [Стр.422]
По характеру гипербилирубинемии выделяют пигментные гепатозы с непрямой (нсконъюгированной) и прямой (конъюгированной)… [Стр.125]
Смотреть другие источники с термином Гепатозы пигментный: [Стр.428] [Стр.
 430]
[Стр.458]
[Стр.459]
[Стр.459]
[Стр.684]
[Стр.610]
[Стр.565]
[Стр.393]
[Стр.51]
[Стр.53]
[Стр.53]
[Стр.244]
[Стр.297]
[Стр.322]
[Стр.566]
[Стр.19]
[Стр.422]
[Стр.427]
[Стр.387]
[Стр.125]
[Стр.729]
[Стр.24]
[Стр.79]
[Стр.125]
[Стр.125]
[Стр.126]
[Стр.127]
[Стр.127]
[Стр.128]
[Стр.132]
[Стр.137]
[Стр.825]
[Стр.414]
[Стр.32]
[Стр.195]
[Стр.124]
[Стр.151]
[Стр.114]
[Стр.507]
430]
[Стр.458]
[Стр.459]
[Стр.459]
[Стр.684]
[Стр.610]
[Стр.565]
[Стр.393]
[Стр.51]
[Стр.53]
[Стр.53]
[Стр.244]
[Стр.297]
[Стр.322]
[Стр.566]
[Стр.19]
[Стр.422]
[Стр.427]
[Стр.387]
[Стр.125]
[Стр.729]
[Стр.24]
[Стр.79]
[Стр.125]
[Стр.125]
[Стр.126]
[Стр.127]
[Стр.127]
[Стр.128]
[Стр.132]
[Стр.137]
[Стр.825]
[Стр.414]
[Стр.32]
[Стр.195]
[Стр.124]
[Стр.151]
[Стр.114]
[Стр.507]
| Клиническая медицина №7 2009 Циммерман Я. С. Контактная информация: Наследственные пигментные гепатозы (функциональные гипербилирубинемии) В обзоре представлены современные данные об обмене билирубина, дефиниция и классификация наследственных пигментных гепатозов, их патогенез и формы наследования, клиническая картина и возможности лабораторной, инструментальной и дифференциальной диагностики, морфологические изменения в печеночной ткани и существующие методы лечения. Обсуждены спорные терминологические вопросы. Ключевые слова: функциональная гипербилирубинемия, диагностика, лечение ЛИТЕРАТУРА 3. Блюгер А. Ф., Крупникова Э. З. Наследственные пигментные гепатозы (диагностика и лечение). Клин. мед. 1984; 4: 137—145. 4. Циммерман Я. С., Пыстогова А. В., Кунстман Т. Г. Синдром Жильбера: опыт динамического наблюдения за диспансерной группой. Клин. мед. 1992; 5—6; 60—63. 5. Маев И. В., Орлов Л. Л., Овчинникова Н. И., Черемушкин С. В. Доброкачественные гипербилирубинемии. Клин. мед. 1999; 6: 9—14. 6. Strassburg C. P., Nguyen N., Manus M. P., Tykey R. H. UDP-glucuronosyltransferase activity in human liver and colon. Gastroenterology 1999; 116 (1): 149—160. 7. Yamamoto K., Sato H., Fujiama Y. et al. Contribution of two missens mutations (G71R and Y486D) of the bilirubin UDP-glucuronosyltransferase (UGT1A1) gene to phenotypes of Gilbert’s syndrome and Crigler—Najjar syndrome type-II. Biochim. Biophys. Acta 1998; 1406 (3): 267—273. 8. Tiribelli C., Bellantini S., Lunazzi G. C., Sottocasa G. L. Role and nature of plasma membrane carrier proteins in the hepatic transport of organic anions.  9. Iyer L., King C. D., Whiginton P. F. Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite in human livers microsomes. J. Clin. Invst. 1998; 101 (4): 847—853. 10. Шерлок Ш., Дули Дж. Заболевания печени и желчных путей: Пер. с англ. М.; 1999. 11. Кузнецов А. С., Фомина И. Г., Тарзиманова А. И., Оганесян К. А. Дифференциальная диагностика доброкачественых гипербилирубинемий. Клин. мед. 2001; 3: 8—13. 12. Гончарик И. И., Кравченко Ю. С. Синдром Жильбера: патогенез и диагностика. Клин. мед. 2001; 4: 40—44. 13. Подымова С. Д. Болезни печени: Руководство для врачей. М.: 2005. 14. Мадьяр И. Заболевания печени и желчных путей: Пер. с венг. Будапешт; 1962; т. 1—2. 15. Губергриц Н. Б. Функциональные гипербилирубинемии. Doctor 2004; 3: 19—22. 16. Gilbert A., Castaigne J., Lereboullet P. De l’ictere familial contribution a l’etude de la diathese biliare.  Bull. Soc. Med. Hцp. (Lyon)
1900; 17: 948—959. Bull. Soc. Med. Hцp. (Lyon)
1900; 17: 948—959.17. Gilbert A., Lereboullet P. La cholemie simple familiale. Sem. Med. (Paris) 1901; 21: 241—243. 18. Clarhe D. J., Moghrabi N., Monaghan G. et al. Genetic defect of the UDP-glucuronosyltransferase-1 gene that cause familial non-gemolytic unconjugated hyperbilirubinaemies. Clin. Chim. Acta 1997; 266 (1): 63—74. 19. Bosma P. J., Chowdhury J. R. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase-1 in Gilbert’s syndrome. N. Engl. J. Med. 1995; 333 (18): 1171—1175. 20. Chalasani N., Chowdhury N. P., Chowdhury J. R., Boyer T. D. Kernicterus in der adult who is heterozygous for Gilbert-type genetic defect. Gastroenterology 1997; 112: 2099—2103. 22. Kowai O., Nishizawa M., Hasada K. et al. Gilbert’s syndrome in caused by a heterozygous missense mutation in the gene for bilirubin UDP-glucuronosyltrasferase.  Hum. Mol. Genet 1995; 4 (7): 693—697. Hum. Mol. Genet 1995; 4 (7): 693—697.23. Sato H., Adachi Y., Kowai O. Genetic basis of Gilbert’s syndrome. Lancet 1996; 347 (9001): 558—565. 24. Алексеев В. Г., Каличенок В. М., Уржумов В. Д., Басос С. Ф. Неконъюгированная гипербилирубинемия у лиц молодого возраста. Тер. арх. 1992; 2: 15—18. 25. Орлов В. А., Барханова А. Г., Захарова Г. Ю., Хруцкая М. С. Диагностика пигментных гепатозов. Рос. мед. журн. 1992; 4: 22—24. 26. Managhan G., Ryan M. Genetic variation in bilirubin UDP-glucuronosyltranferase gene promotor and Gilbert’s syndrome. Lancet 1996; 347 (9001): 578—581. 27. Bosma P. J., Chowdhury J. R., Bakker C. et al. The genetic basis of reduced expression of bilirubin UDP-glucuronosyltrasferase-1 in Gilbert’s syndrome. N. Engl. J. Med. 1995; 333 (5): 1171—1174. 28. Еселев М. М., Сцепуро П. Г. Клиническая картина и диагностика синдрома Жильбера. Сов. мед. 1990; 9: 56—60. 29. Bancroft J. D., Kreamer B. Gilbert’s syndrome accelerates development of neonatal jaundice.  J. Pediatr. 1998; 132 (4): 323—325. J. Pediatr. 1998; 132 (4): 323—325.30. Okobicsanui L., Nassuatto G., Strazzabosco M. et al. Unconjugated hyperbilirubinemia. Clinical, laboratory and metabolic aspects. Z. Gastroenterol. 1993; 31 suppl. 2): 78—80. 31. Лермонтов М. Ю. Герой нашего времени. Собр. соч. в 4-х т. М.; 1976; т. 4: 108—109. 32. Martinez-Bruna M. S. The rifampicin test in the diagnosis of Gilbert syndrome. Aten. Primeria 1993; 11 (2): 84—86. 34. Виноградов А. В. Дифференциальный диагноз внутренних болезней. М.; 1987. 35. Майер К.-П. Гепатит и последствия гепатита: Пер. с нем. М.; 1999. 36. Crigler J. F., Najjar U. A. Congenital familial non-hemolytic jaundice with kernicterus. Pediatrics 1952; 10: 169—175. 37. Ritter J. K., Yeatman M. T., Kaiser C. et al. A phenylalanine codon deletion at the UGT-1 gene compex locus of a Crigler—Najjar type-I patient generates a pH-sensitive bilirubin UDP-glucuronosyltransferase.  J. Biol.
Chem. 1993; 286 (31): 23573—23579. J. Biol.
Chem. 1993; 286 (31): 23573—23579.38. Labrune P., Myara A., Hadchouel M. et al. Genetic heterogenity of Crigler—Najjar syndrome type-I: A study of 14 cases. Hum. Genet. 1994; 94 (6): 693—697. 39. Seppen J., Bosma P. J., Goldhoorn B. G. et al. Discrimination between Crigler—Najjar type-I and -II by expression of mutant bilirubin uridin-diphosphate-glucuronosylotransferase. J. Clin. Invest. 1994; 94 (6): 2385—2391. 40. Fox I. J., Chowdhury J. R., Kautman S. S. et al. Treatment of the Crigler—Najjar syndrome type-I with hepatocyte transplantation. N. Engl. J. Med. 1998; 338 (20): 1422—1426. 41. Gantlae S., Banker C. T. Splice-site mutation: A novel genetic mechanism of Crigler—Najjar syndrome type-I. Am. J. Hum. Genet. 1998; 32 (3): 585—592. 42. Rubbeli G., Ronshi F. A neurophysiological study in children with Crigler—Najjar syndrome type-I. Neuropediatrics 1997; 28 (5): 281—286. 43. Guldutuna S., Landenbck U., Bock K. W. et al. Crigler—Najjar syndrome type-II.  New observation of passible autosomal recessive inheritance. Dig.
Dis. Sci. 1995; 40 (1): 28—32. New observation of passible autosomal recessive inheritance. Dig.
Dis. Sci. 1995; 40 (1): 28—32.45. Luceu J. F., Arias I. M., McKay R. J. Transient familial neonatal hyperbilirubinemia. Am. J. Dis. Child. 1960; 100: 787—792. 46. Dubin I. N., Johnson F. B. Chronic idiopatic jaundice with unidentified pigment in liver cells; new clinicopathologic entity with report of 12 cases. Medicine (Baltimore) 1954; 33: 155—161. 47. Dubin I. N. Chronic idiopathic jaundice: review of 50 cases. Am. J. Med. 1958; 24: 268—274. 48. Бондарь З. А. Клиническая гепатология. М.; 1970. 49. Houbal V. Beitrag zum Dubin—Johnson syndrome. Z. Ges. Inn. Med. 1961; 16: 905—906. 50. Zimniak P. Dubin—Johnson and Rotor syndromes: Molecular basis and pathogenesis. Semin. Liver Dis. 1993; 13 (3): 248—260. 51. Rotor A. B., Manahan L. 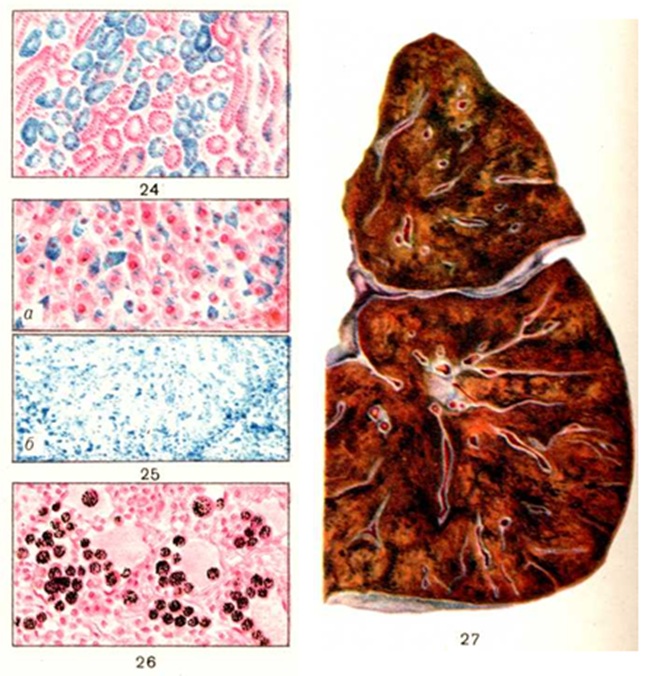 , Florentin A. Familial non-hemolytic jaundice
with direct Van den Bergh reaction. Acta Med. Philipp. 1948; 5: 37—42. , Florentin A. Familial non-hemolytic jaundice
with direct Van den Bergh reaction. Acta Med. Philipp. 1948; 5: 37—42.52. Abei M., Matsuzahi Y., Yanahi N. Defective hepatic glutatione-S-transferase in Rotor’s syndrome. Am. J. Gastroenterol. 1995; 90 (4): 681—684. 53. Chuo Y. N., Chang M. H. Rotor’s syndrome: Report of one case. Acta Pediatr. Sin. 1992; 33 (6): 435—440. 54. De Vos R., De Walf-Peters C., Desmet V. et al. Progressive intrahepatic cholestasis (Byler’s disease): Case report. Gut 1975; 16: 943—947. 55. Carlton V. E. H., Knisely A. S., Freimer N. B. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum. Mol. Genet. 1995; 4: 1049—1054. 56. Williams R., Cartter M. A., Sherlock S. et al. Idiopathic recurrent cholestasis: A study of the functional and pathological lesions in four cases. Quart. J. Med. 1964; 33: 381—391. 57. Brenard R., Geubel A.  P., Benhamou J.-P. Benign recurrent intrahepatic
cholestasis. J. Clin. Gastroenterol. 1989; 11: 546—552. P., Benhamou J.-P. Benign recurrent intrahepatic
cholestasis. J. Clin. Gastroenterol. 1989; 11: 546—552.58. Everson G. T., Ahnen D., Harper P. C. et al. Benign recurrent intrahepatic cholestasis: Treatment with 5-ademethionin. Gastroenterology 1989; 96: 1354—1359. 59. Саркисов Д. С. Следует, наконец, отказаться от понятий «функциональная болезнь», «функциональная патология». Клин. мед. 1998; 3: 64—66. 60. Василенко В. Х. Введение в клинику внутренних болезней. М.; 1985. 13, 20. |
 4
4
Гепатоз — Википедия
Гепатозы — это группа заболеваний печени, в основе которых лежит нарушение обмена веществ в печёночных клетках (гепатоцитах) и развитие в клетках печени дистрофических изменений. При этом воспалительные явления отсутствуют или слабо выражены.[1]
Различают жировые и пигментные гепатозы, имеющие острое и хроническое (чаще) течение. Наиболее распространённым гепатозом является хронический стеатогепатоз (жировой гепатоз, жировая инфильтрация печени, жировая дистрофия печени).
Различают экзогенные и наследственные факторы развития гепатозов. К экзогенным относятся токсические воздействия (острые и хронические), патология других органов и систем и алиментарные (пищевые) факторы — заболевания щитовидной железы, сахарный диабет, ожирение, синдром Кушинга, дефицит белка, авитаминозы и др. Пигментные гепатозы обусловлены нарушениями обмена билирубина и жёлчных кислот в печени.
Жировой гепатоз[править]
Стеатогепатоз — это заболевание печени, сопровождающееся ожирением печёночных клеток. Причины ожирения клеток печени — чаще всего избыточное поступление жиров и углеводов в пищу или их избыточное накопление в крови, вследствие различных нарушений обмена веществ, метаболического синдрома, эндокринные заболевания, воздействие токсических для печени веществ, в том числе алкоголя и гепатотоксических лекарственных средств.
Возможно развитие жирового гепатоза вследствие нарушения выведения жира из печени. Это происходит при снижении количества веществ, участвующих в переработке жиров (белок, липотропные факторы). Нарушается образование из жиров фосфолипидов, бета-липопротеинов, лецитина, вследствие чего «лишние» свободные жиры откладываются в печеночных клетках.
Нарушается образование из жиров фосфолипидов, бета-липопротеинов, лецитина, вследствие чего «лишние» свободные жиры откладываются в печеночных клетках.
Клинические проявления[править]
Клинические проявления хронического гепатоза на начальных этапах, как правило, минимальны, затем происходит постепенное нарастание явлений печёночной недостаточности.
При ультразвуковой диагностике наблюдается равномерное увеличение печени, диффузное повышение её эхогенности (иногда выраженное), с сохранением её однородности (хотя при прогрессировании процесса появляется характерная «зернистость» паренхимы, свидетельствующая о начале развития стеатогепатита и гепатита) и т. д.
При компьютерной томографии выявляется в разной степени выраженное диффузное снижение денситометрических показателей паренхимы печени (ниже 55 HU, иногда вплоть до отрицательных значений, соответствующих плотности жира), как правило отмечается увеличение размеров органа. Возможно выявление ограниченных участков жировой инфильтрации, окружённых неизменённой тканью печени. Чаще локальная жировая инфильтрация наблюдается в S4 печени, имеет довольно ровные, прямые контуры, ход сосудов в инфильтрированной жиром ткани не изменён, масс-эффект (объёмное воздействие на окружающие структуры) отсутствует.
Чаще локальная жировая инфильтрация наблюдается в S4 печени, имеет довольно ровные, прямые контуры, ход сосудов в инфильтрированной жиром ткани не изменён, масс-эффект (объёмное воздействие на окружающие структуры) отсутствует.
Назначается диета с повышенным содержанием белков (не менее полугода), ограничением жиров, особенно тугоплавких животного происхождения. Назначаются препараты улучшающие обмен жиров — витамин B12, фолиевая кислота, липоевая кислота, холин-хлорид, препараты печени — прогепар, рипазон, сирепар. Рекомендуется достаточная физическая активность.
При исключении действия повреждающего фактора и своевременном лечении возможно выздоровление, однако гепатоз в отдельных случаях может трансформироваться в хронический гепатит и цирроз.
Смотрите также[править]
- Алмазов В.А. и др. Клиническая патофизиология. — 2-е издание исправленное и переработанное. — Москва: ВУНМЦ, 1999 год. — 303 с.
| Гепатозы Группа заболеваний печени, сопровождающаяся дистрофией паренхимы без выраженной мезенхимально-клеточной |
 К этой группе относят острые и хронические гепатозы, а также гепатоз беременных.
К этой группе относят острые и хронические гепатозы, а также гепатоз беременных. 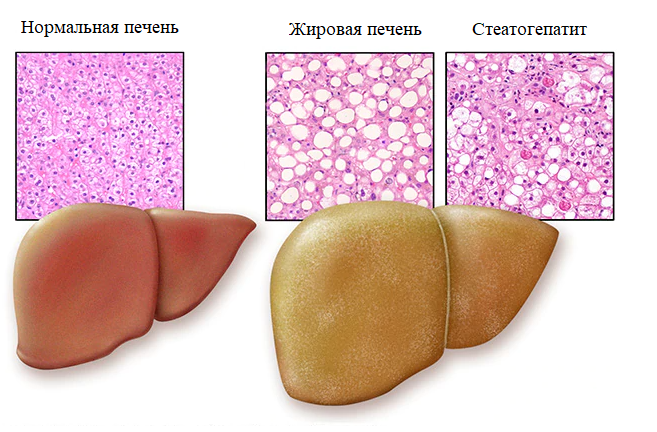 Прогноз тяжелый, в лучшем случае возможен переход в
Прогноз тяжелый, в лучшем случае возможен переход в  Встречается довольно часто, развивается под воздействием алкоголя, токсических веществ
Встречается довольно часто, развивается под воздействием алкоголя, токсических веществ  Однако решающее значение в диагностике
Однако решающее значение в диагностике 
 Субиктеричность склер и кожных
Субиктеричность склер и кожных 
 Заболевание передается по
Заболевание передается по 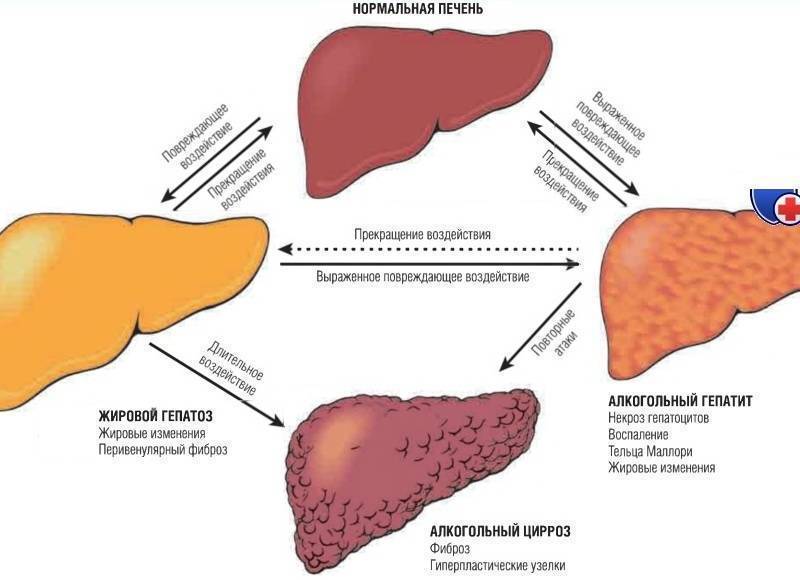 Продолжается она неопределенное время и связана с увеличением неконъюгированного
Продолжается она неопределенное время и связана с увеличением неконъюгированного  Напротив, введение
Напротив, введение  Конъюгационная функция гепатоцитов при этом сохранена. Заболевание предположительно передается по
Конъюгационная функция гепатоцитов при этом сохранена. Заболевание предположительно передается по 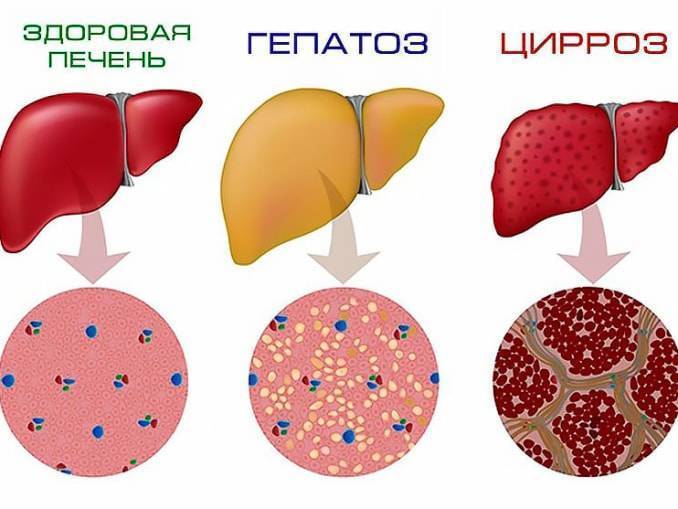

 Печень микроскопически не
Печень микроскопически не 
 Имеется выраженная анемия,
Имеется выраженная анемия, Молекулярно-генетическая верификация нарушений пигментного обмена
Доброкачественные гипербилирубинемии (пигментные гепатозы) – заболевания, связанные с генетически обусловленными дефектами ферментных систем печени, участвующих в конъюгации, транспортировке и экскреции билирубина, проявляющиеся хронической или перемежающейся желтухой, без выраженного нарушения функции и структуры печени и явных признаков гемолиза и холестаза.
Диагностика пигментных гепатозов нередко вызывает определенные трудности у врача и в подобных случаях, пациенты с врожденными формами гипербилирубинемий длительное время необоснованно наблюдаются по поводу хронического гепатита, холестатических заболеваний или гемолитических анемий. Поэтому не верная и не своевременная диагностика доброкачественных гипербилирубинемий ведет к необоснованным затратам времени и средств, в связи с многочисленными и не нужными обследованиями и лечением пациентов.
К доброкачественным гипербилирубинемиям относятся синдромы: Криглера-Найяра I и II типов, Жильбера, Дабина-Джонсона, Ротора, Люси-Дрисколл, Аагенеса, болезнь Байлера и доброкачественный семейный возрастной холестаз. Возникновение всех синдромов обусловлено нарушением обмена билирубина проявляется увеличением в крови уровня неконъюгированного билирубина и накоплением его в тканях. Причиной повышения уровня непрямого билирубина является дефект фермента уридиндифосфатглюкоронилтрансферазы (УДФГТ), участвующего в захвате клетками печени билирубина и связывании его с глюкуроновой кислотой.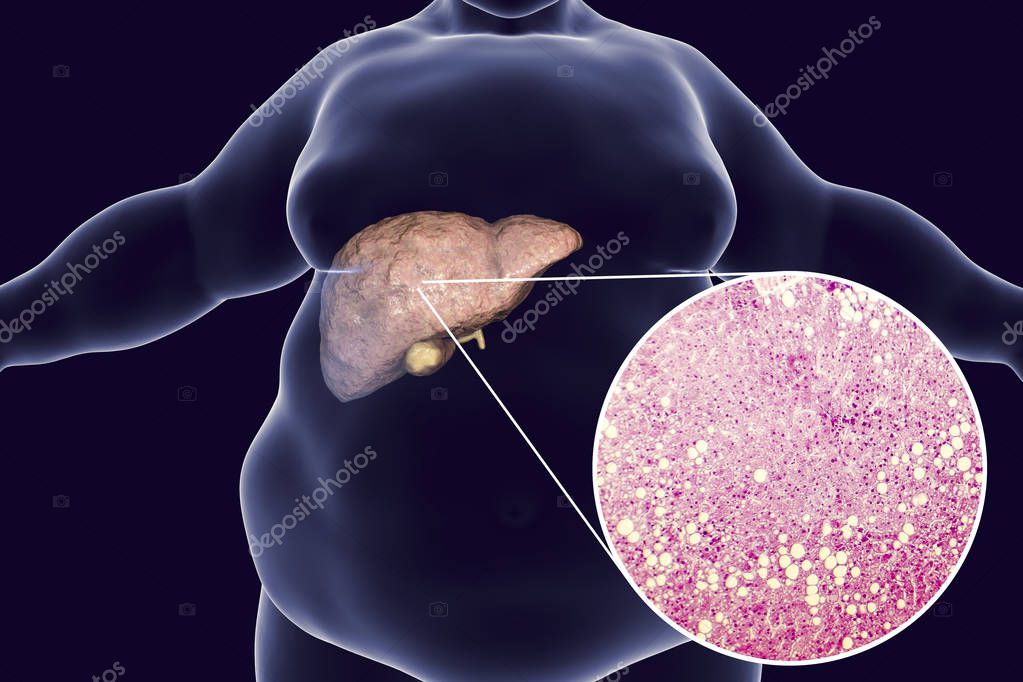 Реакция конъюгации билирубина имеет огромный биологический смысл, превращая высокотоксичный билирубин в малотоксичное, хорошо растворимое соединение билирубин-диглюкуронид (конъюгированный билирубин). Свободный билирубин легко соединяется с эластической тканью, в большом количестве содержится в коже, слизистых, стенках кровеносных сосудов, обусловливая желтуху.
Реакция конъюгации билирубина имеет огромный биологический смысл, превращая высокотоксичный билирубин в малотоксичное, хорошо растворимое соединение билирубин-диглюкуронид (конъюгированный билирубин). Свободный билирубин легко соединяется с эластической тканью, в большом количестве содержится в коже, слизистых, стенках кровеносных сосудов, обусловливая желтуху.
В происхождении кратковременных, транзиторных гипербилирубинемий важную роль играют интоксикации алкоголем и лекарствами (парацетамол, сульфаниламиды, глюкокортикоиды, кофеин и др.). Другой причиной транзиторных гипербилирубинемий у практически здоровых людей являются различные интеркуррентные инфекции (пародонтальные гранулемы, хронический тонзиллит и др.). Несколько реже среди причин оказываются преходящие нарушения кровообращения после физических нагрузок или травм.
Среди доброкачественных гипербилирубинемий самой распространенной формой является синдром Жильбера. Частота его выявлений среди европейцев составляет 2-5%, в США – 3-7%, а среди жителей стран Азии – 3%. Остальные гипербилирубинемии встречаются очень редко; поэтому точных статистических данных о частоте этих синдромов нет.
Остальные гипербилирубинемии встречаются очень редко; поэтому точных статистических данных о частоте этих синдромов нет.
Синдром Жильбера передается по аутосомно-доминантному или аутосомно-рецессивному типу, возраст манифестации приходится на период от 7 до 30 лет и чаще встречается среди лиц мужского пола.
Течение данного заболевания длительно может протекать латентно, являясь случайной находкой при обследовании по поводу другой патологии. Приблизительно у 1/3 больных жалобы отсутствуют, а у 2/3 при тщательном опросе отмечаются астенические и диспепсические явления (подавленное настроение, утомляемость, плохой сон, головокружение тошнота, горечь во рту, снижение аппетита, отрыжка, вздутие живота).
Основной признак болезни – умеренная гипербилирубинемия преимущественно или исключительно за счет увеличения содержания неконъюгированного (непрямого) билирубина. Чаще колебания уровня общего билирубина наблюдаются в пределах 25,6-39,8 мкмоль/л. Существенно реже встречаются повышения уровня билирубина до 119,7-136,8 мкмоль/л. Обычно такие подъемы наблюдаются во время интеркуррентных инфекций, после операций, травм, употребления больших количеств алкоголя. Цитолитические ферменты сыворотки крови (АЛТ, АСТ) в период ремиссии держатся в пределах нормальных значений. В период обострения заболевания приблизительно у 1/4 больных отмечаются небольшое (не более 25-75%) повышение активности аминотрансфераз.
Обычно такие подъемы наблюдаются во время интеркуррентных инфекций, после операций, травм, употребления больших количеств алкоголя. Цитолитические ферменты сыворотки крови (АЛТ, АСТ) в период ремиссии держатся в пределах нормальных значений. В период обострения заболевания приблизительно у 1/4 больных отмечаются небольшое (не более 25-75%) повышение активности аминотрансфераз.
Исключить синдром Жильбера помогают отрицательные результаты провокационных тестов (проба с никотиновой кислотой, фенобарбиталом, с ограничением энергетической ценности пищи).
В настоящее время разработан метод генетического анализа полиморфизма промоторного участка гена UGT1A1 (UDP-glycosyltransferase 1 family, polypeptide A1 gene) кодирующего фермент УДФГТ, основанного на том, что увеличение числа тандемных (ТА) повторов в промоторном участке гена до 7 или 8 (в норме 6) свидетельствует о снижении функциональной активности UGT1A1 и обнаружение подобной мутации позволяет однозначно свидетельствовать о наличии у обследуемого Синдрома Жильбера.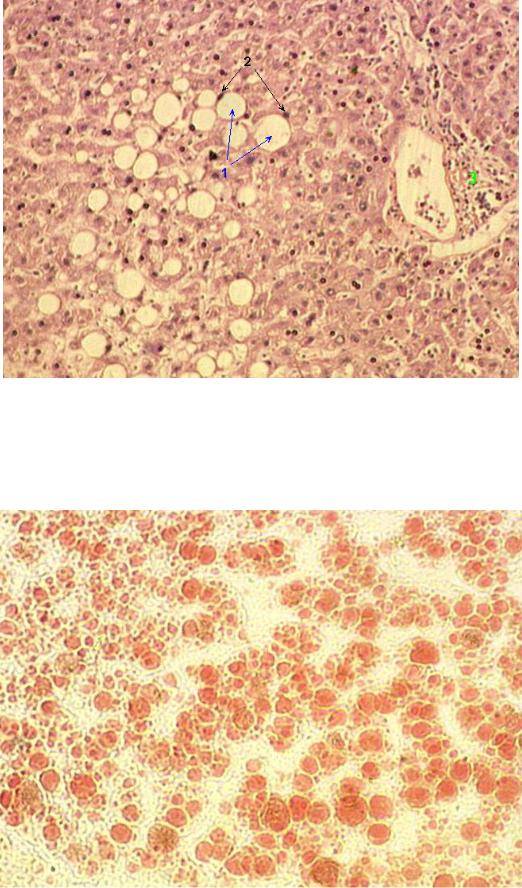
Таким образом, однократное выполнение молекулярно-генетического исследования позволяет установить окончательный диагноз и избежать в дальнейшем многократного проведения у данной категории пациентов многочисленных и рутинных методик верификации нарушений пигментного обмена.
Список литературы.
1. Подымова С.Д. Болезни печени. М.: Медицина, 2005. 768 с.
2. Губергриц Н.Б. Функциональные гипербилирубиемии. М.: Форте принт, 2013. 20 с.: ил.
3. Кузнецов А.С., Фомина И.Г., Тарзиманова А.И., Оганесян К.А. Дифференциальная диагностика доброкачественных гипербилирубиемий // Клиническая медицина. 2001. № 3. С. 8-13.
4. Ройтберг Г.Е. Внутренние болезни. Печень, желчевыводящие пути, поджелудочная железа: учебное пособие/ Под ред. Г.Е. Ройтберг, А.В. Струтынский. М.: МЕДпресс-информ, 2013. 632 с.: ил.
5. Блюгер А.Ф. Наследственные пигментные гепатозы. Л.: Медицина, 1975. 134 с.
6. Окороков А.Н. Диагностика болезней внутренних органов: Т. 1. Диагностика болезней органов пищеварения: М.: Мед. лит., 2000. 560 с.: ил.
1. Диагностика болезней органов пищеварения: М.: Мед. лит., 2000. 560 с.: ил.
Статья добавлена 14 января 2020 г.
Гепатозы пигментные у детей — Педиатр.UA
(синонимы: доброкачественные гипербилирубинемии, семейная негемолитическая желтуха, ювенильная интермиттирующая желтуха, простая семейная холемия). Выявляются в детском и юношеском возрасте, чаще у лиц мужского пола.
Этиология и патогенез. В основе заболевания лежат генетически обусловленные ферментопатии, в итоге которых наблюдаются нарушения внутрипеченочного обмена билирубина. По механизму нарушения билирубинового обмена различают следующие формы патологии.
Мейленграхта-Жильбера симптомокомплекс. Семейная форма заболевания, чаще выявляется в школьном возрасте. Наследование случается по доминантному признаку. Патология характеризуется повышением в крови уровня непрямого билирубина. В норме непрямой билирубин, поступая в печеночную клетку, связывается под влиянием фермента глюкоронилтрансферазы с глюкуроновой кислотой. Образованный прямой билирубин ч/з билиарный конец гепатоцита экскретируется в желчные пути. При снижении глюкоронилтрансферазной активности процесс конъюгации билирубина нарушается и в связи с этим выделяется непрямой билирубин.
Образованный прямой билирубин ч/з билиарный конец гепатоцита экскретируется в желчные пути. При снижении глюкоронилтрансферазной активности процесс конъюгации билирубина нарушается и в связи с этим выделяется непрямой билирубин.
Клиническая картина. Течение патологии волнообразное. Обострения как правило провоцируются интеркуррентными болезнями, нервной и физической перегрузкой. Наблюдается желтушность склер, временами кожи. В отдельных ситуациях сохраняется нормальный цвет кожи и склер при повышенном уровне билирубина (холемия без желтухи). Общее положение как правило не страдает. Печень и селезенка, обычно, не увеличены. У отдельных детей отмечается незначительное повышение мягкой по консистенции печени, связанное с сопутствующей патологией желчных путей (холецистохолангит, дискинезия желчных путей). Функциональные пробы печени, за исключением умеренного повышения уровня билирубина, у подавляющего количества больных не нарушены. Гистологически структура печени не изменена, наблюдаются только нерезкие дистрофические перемены гепатоцитов.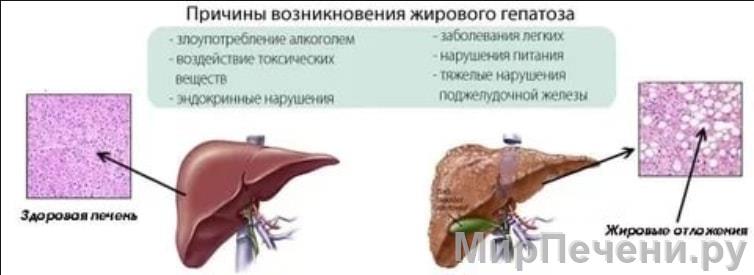
Прогноз. Благоприятный, течение заболевания доброкачественное. Крайним вариантом этой формы патологии является полное или практически полное отсутствие глюкоронилтрансферазы и в итоге — неспособность печени кнъюгировать билирубин (Криглера-Наджара симптомокомплекс). В таких ситуациях желтуха появляется тут же после рождения, носит постоянный характер. Течение патологии тяжелое. В большей части случаев — смертельный исход в раннем детстве.
Дабина-Джонсона и Ротора симптомокомплекс. Характеризуется повышением в крови содержания прямой фракции билирубина, связанным с нарушением экскреции билирубина ч/з мембрану гепатоцита в желчные канальцы. Желтушность и уровень повышения билирубина более выражены, чем при синдроме Мейленграх-таЖильбера. Сильнее страдает состояние ребенка, более выражены общие признаки отравлении. Печень чаще увеличена.
Функциональные пробы как правило нормальные, замедлена только ретенция бромсульфалеина. Гистологически отмечается отложение темных гранул пигмента.
Синдром Гилберта — Better Health Channel
О синдроме Гилберта
При правильном функционировании печень:
- очищает организм от загрязнений и токсинов
- перерабатывает белки и углеводы
- расщепляет жиры с помощью желчи, хранящейся в желчном пузыре.
У человека с синдромом Жильбера печень неспособна последовательно перерабатывать желтовато-коричневый пигмент желчи, называемый билирубином. Это приводит к высокому уровню билирубина в кровотоке, что может вызвать пожелтение кожи и глаз (желтуха).Несмотря на желтушный вид человека, его печень функционирует нормально.
Синдром Жильбера — распространенное легкое заболевание, которое, как считается, передается по наследству примерно в половине всех случаев. Мужчины подвержены более высокому риску, чем женщины, и склонны к развитию синдрома Жильбера в возрасте от позднего подросткового возраста до 30-летнего возраста. Обычно заболевание диагностируется случайно при расследовании не связанных с ним заболеваний.
Обычно заболевание диагностируется случайно при расследовании не связанных с ним заболеваний.
Синдром Жильбера считается безвредным заболеванием и обычно не требует лечения.
Симптомы синдрома Жильбера
Синдром Жильбера обычно не имеет явных симптомов, кроме пожелтения кожи или глаз (желтухи). Иногда сообщается о других симптомах, в том числе:
- Жалобы со стороны желудочно-кишечного тракта
- усталость
- слабость
- боль в животе.
Однако неясно, связаны ли эти симптомы напрямую с повышенным уровнем билирубина.
Синдром Жильбера не связан с вирусным гепатитом, который также вызывает желтуху.У человека с синдромом Жильбера нормальная (соломенная) моча. У человека с гепатитом обычно темная моча, а также может подниматься температура.
Обработка билирубина
Билирубин — желтовато-коричневый пигмент, придающий желчи цвет. Он создается при расщеплении старых красных кровяных телец селезенкой. Дальнейшая переработка билирубина происходит в печени.
Дальнейшая переработка билирубина происходит в печени.
Синдром Жильбера возникает из-за пониженной активности фермента печени, что делает печень менее способной к переработке билирубина.Это приводит к большему, чем обычно, уровню циркуляции билирубина в кровотоке, что может вызвать пожелтение кожи и глаз. У человека с синдромом Жильбера уровень билирубина обычно колеблется и лишь иногда колеблется в пределах нормы.
Диагностика синдрома Жильбера
Во многих случаях синдром Жильбера протекает настолько легко, что нет явных симптомов. Это часто диагностируется случайно, когда назначают анализы крови для исследования несвязанного состояния.В других случаях синдром Жильбера может сопровождаться симптомами, аналогичными более серьезным заболеваниям печени, поэтому необходимо тщательное медицинское обследование.
Диагностические тесты включают:
- история болезни
- медицинский осмотр
- анализов крови
- анализов мочи.

Существует генетический тест, который может обнаружить ген, вызывающий синдром Жильбера, но он обычно не является необходимым для диагностики и широко не доступен.
Лечение не требуется
Синдром Гилберта — это легкое заболевание, которое обычно не требует лечения. Люди с этим расстройством ведут нормальный здоровый образ жизни. Нет никаких доказательств того, что это состояние вредно или ведет к более серьезным заболеваниям.
Некоторые лекарства могут быть повреждены
Иногда наличие синдрома Жильбера может увеличить токсичность некоторых лекарств, используемых для лечения тяжелых заболеваний. Эти препараты включают иринотекан (используется для лечения рака) и индинавир (используется для лечения ВИЧ / СПИДа).Нет никаких доказательств того, что синдром Жильбера влияет на наиболее часто используемые лекарства.
Тем не менее, пациенту с синдромом Жильбера было бы разумно обратиться за дополнительными советами к своему практикующему врачу, прежде чем начинать прием нового лекарства.
Куда обратиться за помощью
Контент-партнер
Эта страница была подготовлена после консультаций и одобрена: Больница Канберры — Гастроэнтерологическое отделение
Последнее обновление: Май 2020 г.
Контент на этом веб-сайте предоставляется только в информационных целях.Информация о терапии, услуге, продукте или лечении никоим образом не поддерживает и не поддерживает такую терапию, услугу, продукт или лечение и не предназначена для замены рекомендаций вашего врача или другого зарегистрированного медицинского работника. Информация и материалы, содержащиеся на этом веб-сайте, не предназначены для использования в качестве исчерпывающего руководства по всем аспектам терапии, продукта или лечения, описанных на веб-сайте. Всем пользователям рекомендуется всегда обращаться за советом к зарегистрированному специалисту в области здравоохранения для постановки диагноза и ответов на свои медицинские вопросы, а также для выяснения того, подходит ли конкретная терапия, услуга, продукт или лечение, описанные на веб-сайте, в их обстоятельствах.Штат Виктория и Департамент здравоохранения и социальных служб не несут ответственности за использование любыми пользователями материалов, содержащихся на этом веб-сайте.
Всем пользователям рекомендуется всегда обращаться за советом к зарегистрированному специалисту в области здравоохранения для постановки диагноза и ответов на свои медицинские вопросы, а также для выяснения того, подходит ли конкретная терапия, услуга, продукт или лечение, описанные на веб-сайте, в их обстоятельствах.Штат Виктория и Департамент здравоохранения и социальных служб не несут ответственности за использование любыми пользователями материалов, содержащихся на этом веб-сайте.
Болезнь печени — обзор
Болезнь Вильсона (гепатолентикулярная дегенерация)
Болезнь Вильсона (БВ) — аутосомно-рецессивное заболевание, которое приводит к аномальному накоплению меди в печени, почках и ЦНС. Одно из старейших заболеваний, признанных семейным, WD впервые было описано Киннером Уилсоном в 1912 году как прогрессирующее заболевание, характеризующееся циррозом печени и размягчением ядра линзообразного ядра. 231 За прошедшее столетие WD превратилась из семейного заболевания со смертельным исходом в заболевание, которое поддается лечению с множеством вариантов лечения. WD присутствует во всех популяциях и встречается примерно у 1 из 30 000 человек. Ответственный ген был расположен в хромосоме 13. Заболевание чаще встречается среди евреев из Восточной Европы и некоторых азиатских популяций. 232
WD присутствует во всех популяциях и встречается примерно у 1 из 30 000 человек. Ответственный ген был расположен в хромосоме 13. Заболевание чаще встречается среди евреев из Восточной Европы и некоторых азиатских популяций. 232
Медь является важным металлом, необходимым для нормального функционирования множества ферментов, включая лизилоксидазу, супероксиддисмутазу, тирозиназу и моноаминоксидазу. 233, 234 Метаболизм меди — сложный процесс. 235 Вкратце, медь всасывается из тонкого кишечника и связывается с альбумином. Печень поглощает более 90% связанного с медью альбумина. 234 В печени медь связывается с апоцерулоплазмином с образованием церулоплазмина. Примечательно, что включение меди в апоцерулоплазмин является АТФ-зависимым процессом. Насыщенный шестью молекулами меди церулоплазмин попадает в кровь.Церулоплазмин также является реактивом острой фазы, повышенные уровни которого обнаруживаются при различных воспалительных состояниях. Во всем организме медь поглощается клетками и доставляется к целевым ферментам после связывания с различными металло-шаперонами, богатыми тиолами. 236 Единственный физиологический способ выведения меди — через желчь. При WD пациенты теряют способность мобилизовать медь для выведения с желчью. 237 Этот дефект приводит к повышению уровня меди в сыворотке крови. Высокий уровень меди вызывает выработку металло-шаперона.Поскольку металло-шапероны способны связывать медь в нетоксичной форме, пациенты обычно остаются бессимптомными до тех пор, пока поступление меди не превысит абсорбционную способность металло-шаперонов. Уровни церулоплазмина низкие у пациентов с WD, как и у пациентов с циррозом печени любой причины. 238
Во всем организме медь поглощается клетками и доставляется к целевым ферментам после связывания с различными металло-шаперонами, богатыми тиолами. 236 Единственный физиологический способ выведения меди — через желчь. При WD пациенты теряют способность мобилизовать медь для выведения с желчью. 237 Этот дефект приводит к повышению уровня меди в сыворотке крови. Высокий уровень меди вызывает выработку металло-шаперона.Поскольку металло-шапероны способны связывать медь в нетоксичной форме, пациенты обычно остаются бессимптомными до тех пор, пока поступление меди не превысит абсорбционную способность металло-шаперонов. Уровни церулоплазмина низкие у пациентов с WD, как и у пациентов с циррозом печени любой причины. 238
Проявления гепатолентикулярной дегенерации широко варьируются. Как правило, у пациентов появляются симптомы заболевания печени до появления неврологических симптомов. 239 Обычно WD проявляется у детей и молодых людей с неспецифическими симптомами, включая тошноту, рвоту и боль в животе. Пациенты могут иметь в анамнезе легкую перемежающуюся желтуху, а у некоторых пациентов может быть гепатомегалия или гепатоспленомегалия. WD может также проявляться как фульминантная печеночная недостаточность 240, 241 или как случайное бессимптомное повышение сывороточных трансаминаз. WD может имитировать различные заболевания печени, включая аутоиммунный гепатит. WD следует учитывать при дифференциальной диагностике установленного заболевания печени даже у ребенка дошкольного возраста. 242
Пациенты могут иметь в анамнезе легкую перемежающуюся желтуху, а у некоторых пациентов может быть гепатомегалия или гепатоспленомегалия. WD может также проявляться как фульминантная печеночная недостаточность 240, 241 или как случайное бессимптомное повышение сывороточных трансаминаз. WD может имитировать различные заболевания печени, включая аутоиммунный гепатит. WD следует учитывать при дифференциальной диагностике установленного заболевания печени даже у ребенка дошкольного возраста. 242
Большинство пациентов обращаются с неврологическими проявлениями WD в зрелом возрасте.Отмечается псевдосклероз, пациенты обращаются с признаками паркинсонизма или ригидной дистонией. Классически описанная линзовидная дегенерация WD обычно проявляется в детстве и чаще связана с дистонией. Детей часто называют «сардонической улыбкой». Клиническим признаком WD является наличие кольца Кайзера-Флейшера, желто-коричневого кольца вокруг роговицы. Кольцо Кайзера-Флейшера вызвано отложением меди в мембране Десцемета. Кольцо лучше всего демонстрируется при исследовании с помощью щелевой лампы; однако кольцо может быть хорошо видно.Кольцо Кайзера-Флейшера присутствует более чем у 98% пациентов с неврологическими проявлениями WD и более чем у 80% пациентов с WD. 243 Примерно 30% пациентов с WD имеют психиатрические симптомы. Наиболее частые симптомы включают депрессию и раздражительность; однако пациенты могут иметь явную кататонию. 243, 244
Кольцо лучше всего демонстрируется при исследовании с помощью щелевой лампы; однако кольцо может быть хорошо видно.Кольцо Кайзера-Флейшера присутствует более чем у 98% пациентов с неврологическими проявлениями WD и более чем у 80% пациентов с WD. 243 Примерно 30% пациентов с WD имеют психиатрические симптомы. Наиболее частые симптомы включают депрессию и раздражительность; однако пациенты могут иметь явную кататонию. 243, 244
Диагностика гепатолентикулярной дегенерации требует высокого индекса подозрительности, потому что проявления похожи на многие причины цирроза.Действительно, WD может лежать в основе сопутствующего заболевания печени. В целом, диагноз следует рассматривать у любого пациента моложе 40 лет с признаками и симптомами печеночной дисфункции, особенно у пациентов с циррозом и необъяснимой дисфункцией ЦНС. Как отмечалось ранее, наличие кольца Кайзера-Флейшера и низкий уровень церулоплазмина в сыворотке практически закрывают диагноз. У пациентов с нормальным уровнем церулоплазмина высокий уровень меди в моче и высокий уровень меди при биопсии печени могут подтвердить диагноз.
У пациентов с нормальным уровнем церулоплазмина высокий уровень меди в моче и высокий уровень меди при биопсии печени могут подтвердить диагноз.
d-Пеницилламин считается «золотым стандартом» в лечении WD, способным обратить вспять печеночные, неврологические и психиатрические проявления.Терапия пеницилламином вряд ли будет эффективной у пациентов с фульминантной недостаточностью, дистонией или тяжелой линзовидной дегенерацией. Побочные реакции на пеницилламин включают сыпь, лимфаденопатию и волчаночный синдром. Опасная для жизни тромбоцитопения 245 или лейкопения 246 встречается редко. Поскольку пеницилламин обладает антипиридоксиновым действием, необходимо принимать пиридоксин. Триентин может быть эффективным у пациентов, чувствительных к пеницилламину. 247, 248 Цинк, который может индуцировать большее производство металло-шаперонов, также эффективен. 249 Трансплантация печени может потребоваться в случае фульминантной печеночной недостаточности и полностью изменит печеночные проявления WD.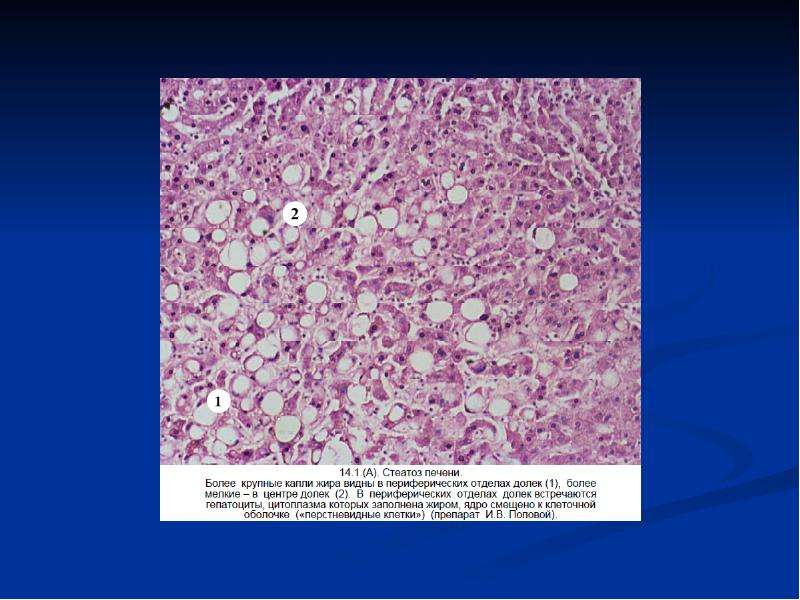 249, 250 Об улучшении неврологических симптомов сообщалось непоследовательно. 249, 251
249, 250 Об улучшении неврологических симптомов сообщалось непоследовательно. 249, 251
Анестезия у пациентов с болезнью Вильсона должна включать в себя тщательную предоперационную оценку в отношении многих систем органов, которые могут быть затронуты. У каждого пациента, принимающего d-пеницилламин, следует определить предоперационный подсчет тромбоцитов.Поскольку метоклопрамид может усугубить экстрапирамидные симптомы пациента, его следует избегать. Дроперидол, прометазин и прохлорперазин также следует избегать у пациентов с гепатолентикулярной дегенерацией, поскольку эти агенты могут усугубить ранее существовавшие двигательные расстройства.
Симптомы, причины, лечение и профилактика
Гепатит — это воспаление печени, вызванное воздействием токсинов, злоупотреблением алкоголем, иммунными заболеваниями или инфекциями. Вирусы вызывают большинство случаев гепатита.
Гепатит А — это тип гепатита, который возникает в результате заражения вирусом гепатита А (HAV). Это острый (краткосрочный) тип гепатита, обычно не требующий лечения.
Это острый (краткосрочный) тип гепатита, обычно не требующий лечения.
По данным Всемирной организации здравоохранения (ВОЗ) ежегодно в мире регистрируется 1,4 миллиона случаев гепатита А. Эта очень заразная форма гепатита может передаваться через зараженную пищу или воду. Как правило, это несерьезно и обычно не вызывает долгосрочных последствий. Инфекция гепатита А обычно проходит сама по себе.
Дети в возрасте до 6 лет обычно не проявляют никаких симптомов заражения вирусом. У детей старшего возраста, подростков и взрослых обычно развиваются легкие симптомы, которые могут включать:
Симптомы обычно появляются через 15–50 дней после заражения вирусом.
У людей развивается гепатит А после заражения ВГА. Этот вирус обычно передается при приеме внутрь пищи или жидкости, загрязненной фекалиями, которые содержат вирус. После передачи вирус распространяется через кровоток в печень, где вызывает воспаление и отек.
Помимо передачи через пищу или питьевую воду, содержащую ВГА, вирус также может передаваться при тесном личном контакте с инфицированным человеком. ВГА заразен, и человек, больной гепатитом А, может легко передать болезнь другим людям, живущим в том же доме.
ВГА заразен, и человек, больной гепатитом А, может легко передать болезнь другим людям, живущим в том же доме.
Вы можете заразиться гепатитом А:
- употребляя пищу, приготовленную кем-то с вирусом гепатита А
- употребляя пищу, которую приготовили приготовители, которые не соблюдают строгие правила мытья рук перед тем, как прикоснуться к еде, которую вы едите
- есть сточные воды зараженные сырые моллюски
- без использования презервативов при половом акте с кем-то, у кого есть вирус гепатита А
- питьевая загрязненная вода
- контакт с фекалиями, инфицированными гепатитом А
Если вы заразитесь вирусом, вы заразитесь за несколько недель до появления симптомов.Заразный период закончится примерно через неделю после появления симптомов.
Гепатит А обычно передается от человека к человеку, что делает его очень заразным. Однако определенные факторы могут увеличить ваш риск заражения, в том числе:
- проживание (или длительное пребывание в) в районе, где распространен гепатит А, включая большинство стран с низкими стандартами санитарии или отсутствием безопасной воды
- инъекционное употребление или употребление запрещенных наркотиков
- проживает в одной семье с лицом, инфицированным гепатитом А
- имеет сексуальную активность с кем-то, кто инфицирован гепатитом А
- является ВИЧ-положительным
По данным ВОЗ, более 90 процентов детей, живущих в странах с низкими санитарными стандартами, к 10 годам заразятся гепатитом А.
После того, как вы обсудите свои симптомы с врачом, он может назначить анализ крови, чтобы проверить наличие вирусной или бактериальной инфекции. Анализ крови покажет наличие (или отсутствие) вируса гепатита А.
У некоторых людей есть только несколько симптомов и никаких признаков желтухи. Без видимых признаков желтухи трудно диагностировать любую форму гепатита с помощью медицинского осмотра. Когда симптомы минимальны, гепатит А может оставаться невыявленным. Осложнения из-за отсутствия диагноза возникают редко.
В очень редких случаях гепатит А может привести к острой печеночной недостаточности. Это осложнение чаще всего встречается у пожилых людей и людей, уже страдающих хроническим заболеванием печени. В этом случае вас госпитализируют. Даже в случае печеночной недостаточности вероятно полное выздоровление. Очень редко требуется пересадка печени.
Не существует формального лечения гепатита А. Поскольку это кратковременная вирусная инфекция, которая проходит сама по себе, лечение обычно направлено на уменьшение симптомов.
Обычно после нескольких недель отдыха симптомы гепатита А начинают улучшаться. Чтобы облегчить симптомы, вам следует:
- избегать алкоголя
- поддерживать здоровую диету
- пить много воды
После отдыха ваше тело, скорее всего, полностью вылечится от гепатита А в течение недель или нескольких месяцев . Обычно заражение вирусом не приводит к долгосрочным негативным последствиям.
После заражения гепатитом А в организме вырабатывается иммунитет к этой болезни.Здоровая иммунная система предотвратит развитие болезни, если вы снова столкнетесь с вирусом.
Способ №1 избежать заражения гепатитом А — это сделать вакцину против гепатита А. Эта вакцина вводится серией из двух инъекций с интервалом от 6 до 12 месяцев.
Если вы путешествуете в страну, где передача гепатита А более распространена, сделайте вакцинацию как минимум за две недели до поездки. Обычно требуется две недели после первой инъекции, чтобы ваш организм начал формировать иммунитет к гепатиту А. Если вы не путешествуете хотя бы год, лучше сделать обе инъекции перед отъездом.
Если вы не путешествуете хотя бы год, лучше сделать обе инъекции перед отъездом.
Проверьте пункт назначения на сайте Центров по контролю и профилактике заболеваний, чтобы узнать, следует ли вам пройти вакцинацию против гепатита А.
Чтобы ограничить вероятность заражения гепатитом А, вам также следует:
- тщательно вымыть руки теплой водой с мылом перед едой или питьем и после посещения туалета
- пить воду в бутылках, а не воду местного производства в развивающихся странах, или в странах с высоким риском заражения гепатитом A
- обедать в известных, уважаемых ресторанах, а не у уличных торговцев
- Избегайте употребления очищенных или сырых фруктов и овощей в районах с низкими санитарными или гигиеническими стандартами
Безалкогольные жирные Заболевание печени — Американский семейный врач
1.Ангуло П. Безалкогольная жировая болезнь печени. N Engl J Med . 2002; 346: 1221–31 ….
2. Кларк Дж. М.,
Diehl AM.
Неалкогольная жировая болезнь печени: малоизвестная причина криптогенного цирроза. ЯМА .
2003. 289: 3000–4.
Кларк Дж. М.,
Diehl AM.
Неалкогольная жировая болезнь печени: малоизвестная причина криптогенного цирроза. ЯМА .
2003. 289: 3000–4.
3. Кольантес Р, Онг JP, Younossi ZM. Безалкогольная жировая болезнь печени и эпидемия ожирения. Клив Клин Дж. Мед . 2004. 71: 657–64.
4. Браунинг Дж. Д., Щепаняк Л.С., Доббинс Р, Нюрнберг П, Хортон Дж. Д., Коэн JC, и другие.Распространенность стеатоза печени среди городского населения США: влияние этнической принадлежности. Гепатология . 2004. 40: 1387–95.
5. Sanyal AJ. Американская гастроэнтерологическая ассоциация. Технический обзор неалкогольной жировой болезни печени AGA. Гастроэнтерология . 2002; 123: 1705–25.
6. Сорби Д.,
Бойнтон Дж.
Lindor KD.
Соотношение аспартатаминотрансферазы и аланинаминотрансферазы: потенциальное значение для дифференциации неалкогольного стеатогепатита от алкогольной болезни печени. Ам Дж. Гастроэнтерол .
1999; 94: 1018–22.
Ам Дж. Гастроэнтерол .
1999; 94: 1018–22.
7. Джозеф А.Е., Саверимутту Ш., аль-Сам С, Повар MG, Максвелл JD. Сравнение гистологии печени с ультрасонографией при оценке диффузного паренхиматозного заболевания печени. Клин Радиол . 1991; 43: 26–31.
8. Hultcrantz R, Габриэльссон Н. Пациенты со стойким повышением аминотрансфераз: обследование с помощью ультразвукового исследования, радионуклидной визуализации и биопсии печени. Дж. Медицинский работник . 1993; 233: 7–12.
9. Сааде С., Юноси З.М., Ремер Э.М., Грамлих Т, Онг JP, Херли М, и другие. Польза радиологической визуализации при неалкогольной жировой болезни печени. Гастроэнтерология . 2002; 123: 745–50.
10. Dienstag JL.
Роль биопсии печени при хроническом гепатите С. Гепатология .
2002; 36 (5 доп. 1): S152–60.
1): S152–60.
11. Angulo P, Кич JC, Баттс КП, Lindor KD.Независимые предикторы фиброза печени у пациентов с неалкогольным стеатогепатитом. Гепатология . 1999; 30: 1356–62.
12. Бедогни Г, Беллентани С. Жирная печень: как часто и почему? Энн Гепатол . 2004; 3: 63–5.
13. Заявление о медицинской позиции Американской гастроэнтерологической ассоциации: неалкогольная жировая болезнь печени. Гастроэнтерология . 2002; 123: 1702–4.
14. Уэно Т., Сугавара Х, Суджаку К., Хашимото О, Цудзи Р, Тамаки С, и другие.Лечебные эффекты ограниченной диеты и физических упражнений у пациентов с ожирением и ожирением печени. Дж. Гепатол . 1997. 27: 103–7.
15. Харрисон С.А.,
Fincke C,
Хелински Д,
Торгерсон С,
Хаяси П.
Пилотное исследование лечения орлистатом пациентов с ожирением и неалкогольным стеатогепатитом. Алимент Фармакол Тер .
2004. 20: 623–8.
Алимент Фармакол Тер .
2004. 20: 623–8.
16. Уйгун А, Кадаифджи А, Исик АТ, Озгуртас Т, Девечи С, Тузун А, и другие.Метформин в лечении больных неалкогольным стеатогепатитом. Алимент Фармакол Тер . 2004; 19: 537–44.
17. Neuschwander-Tetri BA, Брант Э.М., Wehmeier KR, Оливер Д, Бэкон BR. Улучшение неалкогольного стеатогепатита после 48 недель лечения PPAR-гамма-лигандом розиглитазоном. Гепатология . 2003. 38: 1008–17.
18. Промрат К, Лучман Г, Увайфо Г.И., Фридман Р.Дж., Соза А, Хеллер Т, и другие.Пилотное исследование лечения пиоглитазоном неалкогольного стеатогепатита. Гепатология . 2004; 39: 188–96.
19. Басараноглу М,
Акбай О,
Сонсуз А.
Контролируемое испытание гемфиброзила в лечении пациентов с неалкогольным стеатогепатитом. Дж. Гепатол .
1999; 31: 384.
Дж. Гепатол .
1999; 31: 384.
20. Кийджи М, Гюльтен М, Гурель С, Нак СГ, Долар Э, Савчи Г, и другие. Урсодезоксихолевая кислота и аторвастатин в лечении неалкогольного стеатогепатита. Банка Дж Гастроэнтерол . 2003; 17: 713–8.
21. Раллидис Л.С., Дракулис СК, Parasi AS. Правастатин у пациентов с неалкогольным стеатогепатитом: результаты пилотного исследования. Атеросклероз . 2004. 174: 193–6.
22. Линдор К.Д., Каудли К.В., Heathcote EJ, Харрисон МЭ, Йоргенсен Р, Ангуло П, и другие. Урсодезоксихолевая кислота для лечения неалкогольного стеатогепатита: результаты рандомизированного исследования. Гепатология . 2004; 39: 770–8.
23. Lavine JE.
Лечение витамином Е неалкогольного стеатогепатита у детей: пилотное исследование. Дж. Педиатр . 2000; 136: 734–8.
2000; 136: 734–8.
24. Кугельмас М, Hill DB, Вивиан Б, Марсано Л, Макклейн CJ. Цитокины и НАСГ: пилотное исследование влияния изменения образа жизни и витамина Е. Hepatology . 2003; 38: 413–9.
25. Харрисон С.А., Торгерсон С, Хаяси П., Палата J, Шенкер С.Лечение витамином Е и витамином С улучшает фиброз у пациентов с неалкогольным стеатогепатитом. Ам Дж. Гастроэнтерол . 2003. 98: 2485–90.
26. Miglio F, Ровати, г. Санторо А, Сетникар И. Эффективность и безопасность перорального бетаина глюкуроната при неалкогольном стеатогепатите. Двойное слепое рандомизированное проспективное плацебо-контролируемое клиническое исследование в параллельных группах. Arzneimittelforschung . 2000; 50: 722–7.
27.Абдельмалек М.Ф.,
Ангуло П,
Йоргенсен Р.А.,
Сильвестр ПБ,
Lindor KD.
Бетаин, новый многообещающий агент для пациентов с неалкогольным стеатогепатитом: результаты пилотного исследования. Ам Дж. Гастроэнтерол .
2001; 96: 2711–7.
Ам Дж. Гастроэнтерол .
2001; 96: 2711–7.
28. Вайро П., Фонтанелла А, Perna C, Орсо Г, Тедеско М, Де Винченцо А. Устойчивая гипераминотрансфераземия, разрешающаяся после снижения веса у детей с ожирением. Дж. Педиатр .1994; 125: 239–41.
29. Чаласани Н, Aljadhey H, Кестерсон Дж., Мюррей MD, Зал СД. Пациенты с повышенным уровнем печеночных ферментов не имеют повышенного риска гепатотоксичности статинов. Гастроэнтерология . 2004; 126: 1287–92.
30. ЖК Пастернака, Смит СК-младший, Бейрей-Мерц CN, Гранди С.М., Климан Джи, Ленфант С, и другие. ACC / AHA / NHLBI Clinical Advisory по использованию и безопасности статинов. Ход . 2002; 33: 2337–41.
31. Национальная образовательная программа по холестерину. Третий отчет экспертной комиссии по обнаружению, оценке и лечению повышенного холестерина в крови у взрослых. Национальный институт сердца, легких и крови, 2002 г. Доступно онлайн 24 августа 2005 г., по адресу: http://www.nhlbi.nih.gov/guidelines/cholesterol/atp3full.pdf.
Национальный институт сердца, легких и крови, 2002 г. Доступно онлайн 24 августа 2005 г., по адресу: http://www.nhlbi.nih.gov/guidelines/cholesterol/atp3full.pdf.
32. Руссо МВт, Якобсон И.М. Как применять статины пациентам с хроническим заболеванием печени. Клив Клин Дж. Мед .2004. 71: 58–62.
33. Миллер Е.Р. III, Пастор-Барриузо Р., Далал Д, Римерсма РА, Аппель LJ, Гуаллер Э. Мета-анализ: прием высоких доз витамина Е может увеличить общую смертность. Энн Интерн Мед. . 2005; 142: 37–46.
34. Teli MR, Джеймс О.Ф., Берт А.Д., Беннетт МК, День CP. Естественная история неалкогольной жировой дистрофии печени: последующее исследование. Гепатология .1995; 22: 1714–9.
35. Харрисон С.А.,
Торгерсон С,
Хаяси PH.
Естественная история неалкогольной жировой болезни печени: клиническое гистопатологическое исследование. Ам Дж. Гастроэнтерол .
2003. 98: 2042–7.
Ам Дж. Гастроэнтерол .
2003. 98: 2042–7.
36. Адамс Л.А., Lymp JF St, Sauver J, Сандерсон СО, Линдор К.Д., Фельдштейн А, и другие. Естественная история неалкогольной жировой болезни печени: популяционное когортное исследование. Гастроэнтерология . 2005. 129: 113–21.
Обзор болезней печени у крупных животных — пищеварительная система
Клинические признаки заболевания печени могут не проявляться до тех пор, пока> 60–80% паренхимы печени не станет нефункциональной или когда дисфункция печени вторична по отношению к заболеванию другой системы органов. Клинические признаки могут различаться в зависимости от течения заболевания (острого или хронического), первичного очага повреждения (гепатоцеллюлярного, желчного) и конкретной причины. Признаки печеночной энцефалопатии и печеночной недостаточности часто проявляются остро, независимо от того, является ли заболевание печени острым или хроническим. Клинические признаки и тяжесть патологии печени отражают степень нарушения одной или нескольких жизненно важных функций печени, включая регуляцию уровня глюкозы в крови; жировой обмен; производство факторов свертывания крови, альбумина, фибриногена, заменимых аминокислот и белков плазмы; образование и выведение желчи; обмен билирубина и холестерина; преобразование аммиака в мочевину; метаболизм полипептидов и стероидных гормонов; синтез 25-гидроксихолекальциферола; и метаболизм и / или детоксикация многих лекарств и токсинов.
Клинические признаки и тяжесть патологии печени отражают степень нарушения одной или нескольких жизненно важных функций печени, включая регуляцию уровня глюкозы в крови; жировой обмен; производство факторов свертывания крови, альбумина, фибриногена, заменимых аминокислот и белков плазмы; образование и выведение желчи; обмен билирубина и холестерина; преобразование аммиака в мочевину; метаболизм полипептидов и стероидных гормонов; синтез 25-гидроксихолекальциферола; и метаболизм и / или детоксикация многих лекарств и токсинов.
Иктеризация, потеря веса или ненормальное поведение часто встречаются у лошадей с заболеваниями печени и печеночной недостаточностью. Признаки ЦНС часто являются начальным и преобладающим признаком у лошадей с острой печеночной недостаточностью, тогда как потеря веса является заметным признаком у большинства, но не у всех лошадей с хроническим заболеванием печени и недостаточностью. Может присутствовать фотосенсибилизация и, реже, двусторонний паралич глотки, вызывающий инспираторный стридор, диарею или запор. У пораженного крупного рогатого скота обычно наблюдается отсутствие аппетита, снижение надоев молока и потеря веса.Тенезмы и асцит наблюдаются у крупного рогатого скота, но не часто у пораженных лошадей. Похудание может быть единственным признаком абсцессов печени. Иктеричность, которая наиболее выражена при поражении желчевыводящей системы, также часто встречается у лошадей с острой печеночной недостаточностью. В большей степени он присутствует у лошадей с хронической печеночной недостаточностью или у жвачных животных. Гипербилирубинемия натощак является более частой причиной желтухи у лошадей и не связана с заболеваниями печени. Иногда стойкая гипербилирубинемия (прежде всего непрямой или неконъюгированный билирубин) может наблюдаться у здоровых лошадей (особенно чистокровных) без признаков гемолиза или заболевания печени.У жвачных животных желтуха чаще возникает из-за гемолиза и в первую очередь связана с увеличением непрямого билирубина. Гипербилирубинемия, вызванная обструктивными состояниями желчевыводящих путей, редко встречается у коз и овец.
У пораженного крупного рогатого скота обычно наблюдается отсутствие аппетита, снижение надоев молока и потеря веса.Тенезмы и асцит наблюдаются у крупного рогатого скота, но не часто у пораженных лошадей. Похудание может быть единственным признаком абсцессов печени. Иктеричность, которая наиболее выражена при поражении желчевыводящей системы, также часто встречается у лошадей с острой печеночной недостаточностью. В большей степени он присутствует у лошадей с хронической печеночной недостаточностью или у жвачных животных. Гипербилирубинемия натощак является более частой причиной желтухи у лошадей и не связана с заболеваниями печени. Иногда стойкая гипербилирубинемия (прежде всего непрямой или неконъюгированный билирубин) может наблюдаться у здоровых лошадей (особенно чистокровных) без признаков гемолиза или заболевания печени.У жвачных животных желтуха чаще возникает из-за гемолиза и в первую очередь связана с увеличением непрямого билирубина. Гипербилирубинемия, вызванная обструктивными состояниями желчевыводящих путей, редко встречается у коз и овец.![]()
Печеночная энцефалопатия связана с изменениями поведения у лошадей, жвачных животных и свиней. Тяжесть печеночной энцефалопатии часто отражает степень печеночной недостаточности, но не различает острую или хроническую печеночную недостаточность. Признаки печеночной энцефалопатии варьируются от неспецифической депрессии и летаргии до давления на голову, кружения, бесцельной ходьбы, дисфагии, атаксии, дисметрии, постоянной зевоты, пика, повышенного дружелюбия, агрессивности, ступора, судорог или комы.В некоторых случаях печеночной недостаточности, особенно у пони, возникает коллапс глотки или гортани с громкими хриплыми звуками при вдохе и одышкой. Патогенез печеночной энцефалопатии неизвестен, но предлагаемые теории включают аммиак как нейротоксин, изменения в нейротрансмиттерах моноаминов (серотонин, триптофан) или катехоламиновых нейротрансмиттеров, дисбаланс между ароматическими аминокислотами и аминокислотами с короткими разветвленными цепями, что приводит к увеличению тормозящих нейротрансмиттеров (γ-аминомасляная кислота). l-глутамат), нейроингибирование из-за повышенных церебральных уровней эндогенных бензодиазепиноподобных веществ, повышенной проницаемости гематоэнцефалического барьера и нарушения энергетического обмена в ЦНС.Хотя симптомы могут быть очень серьезными, печеночная энцефалопатия потенциально обратима, если можно вылечить основное заболевание печени.
l-глутамат), нейроингибирование из-за повышенных церебральных уровней эндогенных бензодиазепиноподобных веществ, повышенной проницаемости гематоэнцефалического барьера и нарушения энергетического обмена в ЦНС.Хотя симптомы могут быть очень серьезными, печеночная энцефалопатия потенциально обратима, если можно вылечить основное заболевание печени.
Фотосенсибилизацию, которая может проявляться вторично по отношению к острой или хронической печеночной недостаточности, следует дифференцировать от первичной фотосенсибилизации (см. Фотосенсибилизация). Гепатогенная фотосенсибилизация развивается, когда нарушенная функция печени приводит к проникновению филлоэритрина, фотодинамического метаболита хлорофилла в кожу. Филлоэритрин в коже реагирует с ультрафиолетом и высвобождает энергию, вызывая воспаление и повреждение кожи.Признаки фотосенсибилизации разнообразны, но включают беспокойство, боль, зуд, дерматит от легкой до тяжелой степени с эритемой, обширный подкожный отек, изъязвление кожи, шелушение кожи и офтальмию с слезотечением, светобоязнь и помутнение роговицы. Дерматит и отек особенно заметны на непигментированных, светлых или безволосых участках тела и участках, подверженных воздействию солнца. Кожно-слизистые соединения и участки белого волоса являются наиболее частыми участками фотосенсибилизации у крупного рогатого скота. Иногда может быть поражена нижняя часть языка.Возможные последствия — слепота, пиодермия, потеря физического состояния и иногда смерть. Зуд может быть результатом фотосенсибилизации или отложения солей желчных кислот в коже, вторичных по отношению к нарушениям экскреции через печень.
Дерматит и отек особенно заметны на непигментированных, светлых или безволосых участках тела и участках, подверженных воздействию солнца. Кожно-слизистые соединения и участки белого волоса являются наиболее частыми участками фотосенсибилизации у крупного рогатого скота. Иногда может быть поражена нижняя часть языка.Возможные последствия — слепота, пиодермия, потеря физического состояния и иногда смерть. Зуд может быть результатом фотосенсибилизации или отложения солей желчных кислот в коже, вторичных по отношению к нарушениям экскреции через печень.
У животных с заболеванием печени может наблюдаться диарея или запор. Диарея чаще встречается у крупного рогатого скота, чем у лошадей с хроническим заболеванием печени или у животных с хроническим фасциолезом и отравлением гепатотоксическими растениями. У пони и лошадей с гиперлипемией и печеночной недостаточностью могут развиться диарея, ламинит и отек брюшины.У некоторых животных с заболеванием печени диарея и запор чередуются. У лошадей с печеночной недостаточностью и печеночной энцефалопатией часто возникает закупорка толстой кишки из-за уменьшения потребления воды. Запор характерен для отравления Lantana у коз и других жвачных животных.
У лошадей с печеночной недостаточностью и печеночной энцефалопатией часто возникает закупорка толстой кишки из-за уменьшения потребления воды. Запор характерен для отравления Lantana у коз и других жвачных животных.
Рецидивирующие колики, перемежающаяся лихорадка, желтуха, потеря веса и печеночная энцефалопатия могут наблюдаться у лошадей с холелитом, закупоривающим общий желчный проток. Инфекционное или воспалительное заболевание печени или неспособность печени предотвратить попадание эндотоксина в системный кровоток также может привести к перемежающейся лихорадке и коликам.Боль в животе из-за давления на капсулу печени из-за набухания паренхимы часто наблюдается у животных с острым диффузным гепатитом или травмой самой капсулы. Больные животные стоят с изогнутой спиной, не хотят двигаться или проявляют признаки колик. У жвачных животных боль может быть локализована в печени при пальпации по передней вентролатеральной части живота или нескольким последним ребрам с правой стороны. Тенезмы с последующим выпадением прямой кишки наблюдаются у некоторых жвачных животных с заболеваниями печени. Это может быть связано с диареей, печеночной энцефалопатией или отеком кишечника из-за портальной гипертензии.
Тенезмы с последующим выпадением прямой кишки наблюдаются у некоторых жвачных животных с заболеваниями печени. Это может быть связано с диареей, печеночной энцефалопатией или отеком кишечника из-за портальной гипертензии.
Гипоальбуминемия не так часто ассоциируется с заболеванием печени у лошадей, как считалось ранее. Из-за длительного периода полувыведения (~ 19-20 дней у лошадей, ~ 16 дней у коров) и резерва печени для производства альбумина гипоальбуминемия обычно является очень поздним событием в процессе болезни. Концентрации общего белка в сыворотке крови могут быть нормальными или повышенными из-за увеличения β-глобулинов у лошадей с заболеваниями печени. Гипоальбуминемия и гипопротеинемия чаще всего развиваются при хроническом заболевании печени, и они часто встречаются у лам с заболеванием печени.Может возникнуть генерализованный асцит или зависимый отек. Асцит связан с портальной гипертензией, вызванной венозной блокадой и повышенным гидростатическим давлением, а также с утечкой белка в брюшную полость.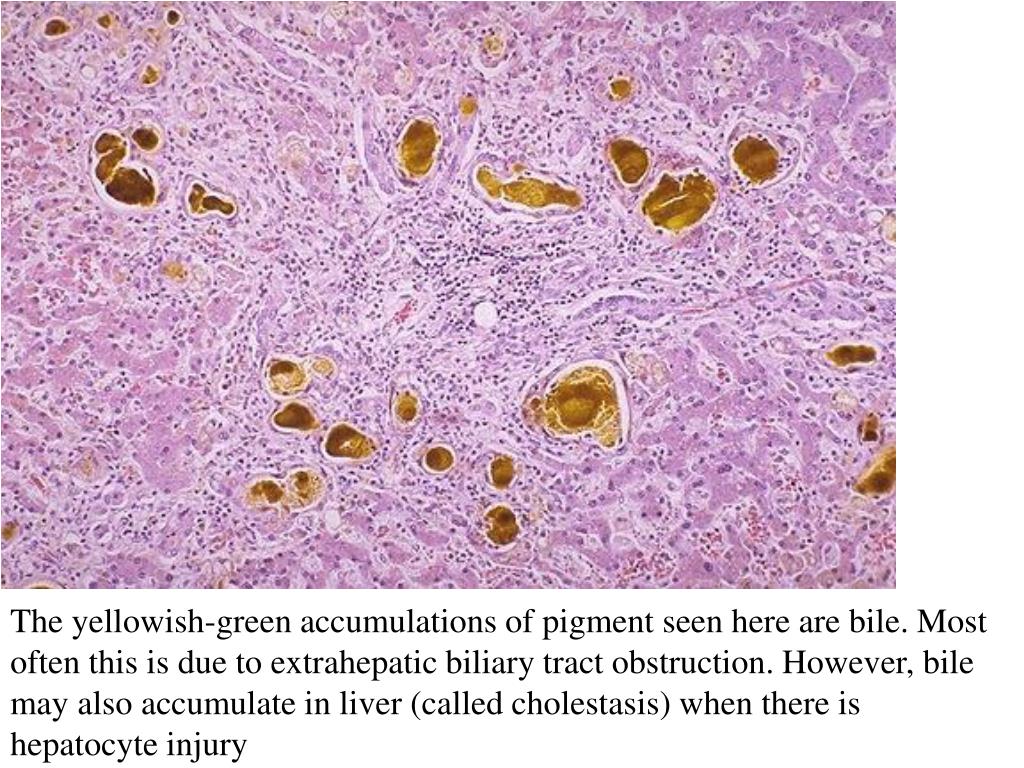 Жидкость в брюшной полости, присутствующая при заболевании печени, обычно представляет собой модифицированный транссудат. Гипоальбуминемия может усугубить асцит, но если она проявляется отдельно, она с большей вероятностью вызовет межнижнечелюстной отек, отек груди или брюшной стенки. Асцит трудно выявить у лошадей и взрослого крупного рогатого скота, если он не обширен.Асцит часто встречается у телят с циррозом печени.
Жидкость в брюшной полости, присутствующая при заболевании печени, обычно представляет собой модифицированный транссудат. Гипоальбуминемия может усугубить асцит, но если она проявляется отдельно, она с большей вероятностью вызовет межнижнечелюстной отек, отек груди или брюшной стенки. Асцит трудно выявить у лошадей и взрослого крупного рогатого скота, если он не обширен.Асцит часто встречается у телят с циррозом печени.
Анемия может наблюдаться у животных с дисфункцией печени из-за паразитарных заболеваний, хронической токсичности меди (у жвачных), отравлений некоторыми растениями или хронических воспалительных заболеваний. Анемия при остром фасциолозе возникает в результате сильного кровоизлияния в брюшную полость, когда личинки проникают в капсулу печени. Травма и кормление взрослых двуусток в желчных протоках вызывают анемию и гипопротеинемию у животных с хроническим фасциолезом.Хроническое воспалительное заболевание (например, абсцессы печени, неоплазия) может вызывать анемию без сопутствующей гипопротеинемии.
Клинические признаки тяжелой или терминальной печеночной недостаточности включают коагулопатии и кровоизлияния из-за снижения выработки факторов свертывания крови печенью и, возможно, повышенной утилизации при септических или воспалительных процессах. Обычно сначала наблюдается удлинение протромбинового времени, потому что фактор VII имеет самый короткий период полувыведения из плазмы. У лошадей может развиться терминальный гемолитический криз, вызванный повышенной хрупкостью эритроцитов.У жвачных животных об этом не сообщалось.
Цвет кала редко меняется у взрослых травоядных с заболеваниями печени. У молодых жвачных и животных с однокамерным желудком холестаз может приводить к выделению более светлых фекалий из-за потери стеркобилина, метаболита билирубина.
Заболевание печени следует всегда рассматривать, если неспецифические клинические признаки, такие как депрессия, потеря веса, перемежающаяся лихорадка и повторяющиеся колики, присутствуют без видимой причины. Различие между острым и хроническим гепатитом или его недостаточностью на основании продолжительности клинических признаков до проявления может вводить в заблуждение, потому что процесс заболевания часто прогрессирует до того, как становятся очевидными клинические признаки. Ранние неясные признаки депрессии и снижения аппетита можно не заметить. Биопсия печени для определения типа патологии, степени фиброза печени и регенеративных возможностей паренхимы печени необходима для разработки плана лечения и получения точного прогноза.
Различие между острым и хроническим гепатитом или его недостаточностью на основании продолжительности клинических признаков до проявления может вводить в заблуждение, потому что процесс заболевания часто прогрессирует до того, как становятся очевидными клинические признаки. Ранние неясные признаки депрессии и снижения аппетита можно не заметить. Биопсия печени для определения типа патологии, степени фиброза печени и регенеративных возможностей паренхимы печени необходима для разработки плана лечения и получения точного прогноза.
Гепатоз: виды и принципы лечения
Гепатозы — группа заболеваний печени, протекающих с нарушением метаболизма гепатоцитов (клеток печени), с развитием дистрофических изменений на фоне слабо выраженных воспалительных изменений или без них.
Основные причины развития гепатозов
Гепатоз может быть врожденным (наследственным) или приобретенным. К наследственным пигментным гепатозам относят наследственное генетически поражение печени — энзимопатии, протекающие с нарушением метаболизма билирубина и периодически проявляющиеся желтухой. Изменения структуры печени при этих типах гепатозов небольшие и имеют вид отложения пигмента.
Изменения структуры печени при этих типах гепатозов небольшие и имеют вид отложения пигмента.
Пигментные гепатозы
- Врожденная негемолитическая желтуха I типа (синдром Криглерара Найяра) — в клетках печени отсутствуют ферменты, связывающие билирубин, поэтому он проникает во все органы в высоких концентрациях, оказывая на них токсическое действие (центральная нервная система, сердце, особенно серьезно поражены мышцы).)
- Врожденная негемолитическая желтуха II типа (синдром Ариаса) — в печени вырабатывается небольшое количество связывающего фермент билирубина, поэтому отмечаются выраженные токсические поражения органов и в первую очередь центральной нервной системы.
- Синдром Жильбера (негемолитическая семейная желтуха) — встречается у 5% обследуемых на желтуху (90% из них мужчины), обычно возникает при периодическом повышении несвязанного (непрямого) билирубина в крови из-за нарушения его проникновения в клетки печени, иногда сочетается с гемолитическим синдромом.
- Синдром Джонсона и синдром Ротора связаны с врожденным дефектом выделительной функции клеток печени. При этом в крови повышается содержание билирубина, а выделение желчных кислот не нарушается.Заболевание впервые проявляется в возрасте 25 лет, чаще у мужчин.
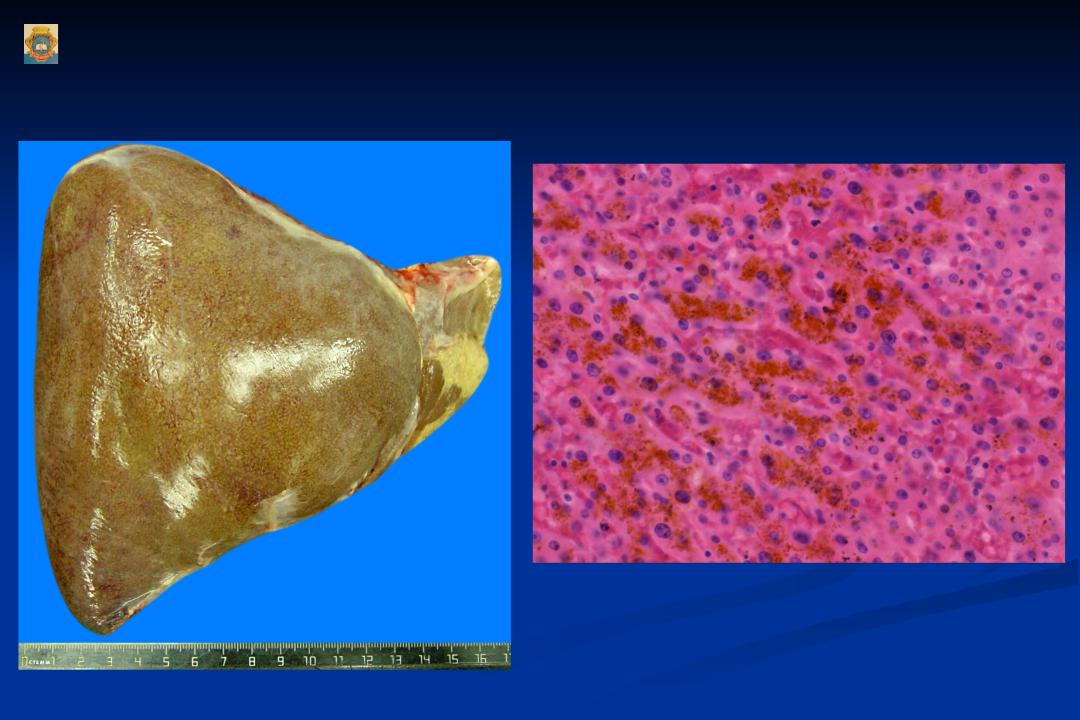
Приобретенный гепатоз
Это группа поражений печени, связанных с развитием дистрофических изменений клеток печени (гепатоцитов), чаще — без грубых структурных изменений, но с длительным течением заболевания, нарушением белкового и других типов обмена в печени, фиброзом. возможно развитие и в тяжелых случаях некроз. Они бывают нескольких типов:
- Острый гепатоз (острая токсическая дистрофия печени) чаще бывает результатом тяжелого токсического отравления (алкоголь и его суррогаты, несъедобные грибы, лекарственные препараты, мышьяк, инсектициды и др.)), а также сепсис, вирусный гепатит В.
- Хронический жировой гепатоз обычно связан с нарушением обмена веществ в клетках печени и отложением в них жира. Чаще всего наблюдается у людей, страдающих общим ожирением (метаболическим синдромом), при несбалансированном избыточном питании, при сахарном диабете, при алкоголизме, отравлениях и т. Д.
- Хронический холестатический гепатоз — может развиться при длительном лечении некоторыми лекарствами (аминазином, аналогами тестостерона, прогестагенами и др.) И является следствием нарушения желчеобразования и холестерина в клетках печени.
 Чаще всего наблюдается у людей, страдающих общим ожирением (метаболическим синдромом), при несбалансированном избыточном питании, при сахарном диабете, при алкоголизме, отравлениях и т. Д.
Чаще всего наблюдается у людей, страдающих общим ожирением (метаболическим синдромом), при несбалансированном избыточном питании, при сахарном диабете, при алкоголизме, отравлениях и т. Д.Основные проявления наследственных пигментных гепатозов.
При синдроме Криглерар-Найяра желтуха развивается сразу после рождения и наблюдается на протяжении всей жизни. При этом у грудничков часто наблюдаются судороги, повышенный мышечный тонус, отставание в умственном и физическом развитии, часто пациенты умирают в детстве из-за поражения нервной системы.
Остальные виды наследственных пигментных гепатозов протекают доброкачественно и не приводят к сокращению продолжительности жизни.Работоспособность незначительно нарушается только в период желтухи, которая может впервые появиться только в подростковом возрасте или у взрослого человека после стресса, ОРЗ и других заболеваний после приема алкоголя. При некоторых синдромах Жильбера первое проявление возникает только после острого вирусного гепатита.
При некоторых синдромах Жильбера первое проявление возникает только после острого вирусного гепатита.
Общее самочувствие больных удовлетворительное, но иногда их беспокоит ощущение тяжести и дискомфорта в правом подреберье (из-за присоединения небольшого воспалительного процесса в желчевыводящих путях), тошнота, горечь в во рту, изжога, потеря аппетита.Может наблюдаться метеоризм, дискомфорт в груди, повышенная утомляемость, апатия, нарушение сна. В то же время желтуха, склера, кожа, слизистые оболочки — главный симптом болезни. Он умеренный, другой может сопровождаться легким кожным зудом, в некоторых случаях появляются небольшие пигментные пятна на лице и веках.
Синдромы Гилберта и диабет Джонсона могут обостряться несколько раз в год, но обычно заканчиваются восстановлением здоровья в течение 2-3 недель.
Основные клинические проявления приобретенных гепатозов.
Острый жировой гепатоз (токсическая дистрофия печени) проявляется выраженными расстройствами пищеварения с быстрым развитием общей интоксикации и желтухи. Уже в начале заболевания печень увеличивается, она становится болезненной, при прогрессировании заболевания может наблюдаться развитие тяжелой печеночной недостаточности, появление кровоизлияния (кровоизлияния) на коже. В тяжелых случаях возможен летальный исход от печеночной комы, а в более легких случаях заболевание переходит в хроническую форму.
Уже в начале заболевания печень увеличивается, она становится болезненной, при прогрессировании заболевания может наблюдаться развитие тяжелой печеночной недостаточности, появление кровоизлияния (кровоизлияния) на коже. В тяжелых случаях возможен летальный исход от печеночной комы, а в более легких случаях заболевание переходит в хроническую форму.
При хронической жировой болезни печени нарушения пищеварения развиваются медленнее, сопровождаются общей слабостью, ноющими болями и чувством вывиха в правом подреберье. Печень постепенно увеличивается в размерах и может быть болезненной при пальпации, но не плотной (как, например, при циррозе). При соответствующем лечении и устранении воздействия всех негативных факторов возможно выздоровление, но в тяжелых случаях возможно развитие хронического гепатита и цирроза печени.
Холестатический гепатоз может протекать как острое заболевание, но чаще принимает длительное хроническое течение.Наиболее характерным для этой формы гепатоза является появление желтухи с сильным зудом и пониженной температурой из-за холестаза (застоя желчи). При этом стул обесцвечивается, а моча приобретает темный цвет. Часто из-за развития вторичного холангита (воспаления желчных протоков) и без должного лечения происходит переход в хронический гепатит
При этом стул обесцвечивается, а моча приобретает темный цвет. Часто из-за развития вторичного холангита (воспаления желчных протоков) и без должного лечения происходит переход в хронический гепатит
Обследование на гепатозы.
- Общий анализ крови и общий анализ мочи,
- Определение билирубина в крови и натощак,
- Проба с фенобарбиталом и проба с никотиновой кислотой,
- Анализ кала на стеркобилин,
- Ферменты крови: АСТ, АЛТ, ГГТФ, ЛФ и другие,
- Белки сыворотки и их фракции
- Протромбиновое время
- Маркеры гепатита В, С, D,
- Тест на бромсульфаз
- УЗИ брюшной полости
- Чрескожная пункционная биопсия печени под контролем УЗИ (по показаниям),
- Генетическое консультирование (при необходимости) и другие исследования и консультации
Основные принципы лечения наследственных гепатозов
Необходимо постоянное соблюдение диеты (таблица № 5 по Певзнеру), адекватный труд и отдых (во избежание стрессов и переутомления), с осторожностью применять лекарственные средства, особенно те, которые могут отрицательно повлиять на работу печени. Желательно периодически проходить курсы гепатопротекторов.
Желательно периодически проходить курсы гепатопротекторов.
Основные принципы лечения приобретенных гепатозов
- При остром токсическом гепатозе необходимо лечение в стационаре. При этом большое внимание уделяется борьбе с геморрагическим синдромом, генерализованным токсикозом, в тяжелых случаях к лечению добавляют кортикостероиды, а также лечению (или профилактике) печеночной недостаточности
- При хроническом гепатозе необходимо купировать действие фактора, приведшего к возникновению заболевания.Необходимо строго придерживаться диеты с ограничением жиров животного происхождения, но с повышенным содержанием полноценного животного белка. Рекомендуется принимать гепатопротекторы — препараты, улучшающие обменные процессы в клетках печени, защищающие их от гибели.
- При холестатическом гепатозе, помимо диеты, часто назначают кортикостероиды, гепатопротекторы.
Витилиго у коровы, очевидно связанное с гепатозом
Yeruham I, Perl S, Gur Y, Avidar YNew Zealand Veterinary Journal , Volume 46, Issue 2, pp 78-79, Apr 1998
Класс статьи: Переписка
Тип животных: Крупный рогатый скот Домашний скот, Производственное животное, Жвачные животные
Тема терминов: Клиническая патология, Диагностические процедуры, Идиопатическое заболевание, Воспаление, Покровы / кожа / шерсть / волосы / мех / перо, Печень / заболевание печени, Травмы / травмы
Издательство: Тейлор и ФрэнсисРеферат
Вилиго — это приобретенная идиопатическая депигментация, при которой отсутствуют предшествующие или сопутствующие признаки кожного воспаления или травмы (Скотт, 1988). О витилиго чаще всего сообщается у голштино-фризской породы (Meijer, van der Ejk, 1961; Meijer, 1965; Meijer, 1966), и клинически он напоминает болезнь арабских лошадей (Scott, 1988). О витилиго также сообщалось у японского черного крупного рогатого скота в сочетании с гипертиреозом (Sako, 1983; Hayashi et al., 1989). Мы сообщаем здесь о случае витилиго у коровы, очевидно связанного с гепатозом. У 3-летней израильской молочной коровы голштинской породы из стада из 40 коров в анамнезе была обнаружена депигментация кожи.Стадо содержится в беспривязном содержании в больших, полностью закрытых открытых сараях, в рамках системы управления с нулевым выпасом. Корова была на 5-м месяце беременности, и за 2 недели до представления наблюдались потеря аппетита и снижение надоев. На клиническом осмотре…
О витилиго чаще всего сообщается у голштино-фризской породы (Meijer, van der Ejk, 1961; Meijer, 1965; Meijer, 1966), и клинически он напоминает болезнь арабских лошадей (Scott, 1988). О витилиго также сообщалось у японского черного крупного рогатого скота в сочетании с гипертиреозом (Sako, 1983; Hayashi et al., 1989). Мы сообщаем здесь о случае витилиго у коровы, очевидно связанного с гепатозом. У 3-летней израильской молочной коровы голштинской породы из стада из 40 коров в анамнезе была обнаружена депигментация кожи.Стадо содержится в беспривязном содержании в больших, полностью закрытых открытых сараях, в рамках системы управления с нулевым выпасом. Корова была на 5-м месяце беременности, и за 2 недели до представления наблюдались потеря аппетита и снижение надоев. На клиническом осмотре…Доступ к полному тексту этой статьи:
через веб-сайт другого провайдера:
Если вы являетесь участником или подписчиком и считаете, что у вас должен быть доступ:
логин
В противном случае:
Зарегистрироваться для учетной записи
Запросить новый пароль
Все литературные материалы Новозеландского ветеринарного журнала принадлежат авторскому праву Тейлор и Фрэнсис .