Нейтрофіли та онкогенез
Резюме. В огляді наведено дані про роль нейтрофілів периферичної крові і нейтрофілів, які інфільтрують пухлини, у протипухлинних та пухлиностимулюючих процесах, які відбуваються при розвитку багатьох видів раку. Наведено характеристику двох основних фенотипів Н1 і Н2 клітин, обговорюються їх функції і механізми переходу від Н1 клітин з протипухлинною активністю в Н2 клітини, які стимулюють проліферацію пухлинних клітин, ангіогенез і метастазування. Наведено дані про функціонально важливі цитокіни та хемокіни, що виділяються пухлинними клітинами, мікрооточенням, лімфоцитами і самими нейтрофілами, які стимулюють вироблення кістковим мозком нейтрофілів і спричиняють накопичення цих клітин у пухлинному вогнищі, розвиток нейтрофілії в крові. Описано основні функціональні молекули нейтрофілів, такі як нейтрофільна еластаза, катепсин, матриксна металопротеїназа-9, аргіназа 1 та ін., з якими зв’язуються протуморогенні властивості Н2 нейтрофілів.
Резюме. В обзоре приведены данные о роли нейтрофилов периферической крови и нейтрофилов, инфильтрирующих опухоли, в противоопухолевых и опухольстимулирующих процессах, которые происходят при развитии многих видов раков. Приведена характеристика двух основных фенотипов Н1 и Н2 клеток, обсуждаются их функции и механизмы перехода от Н1 клеток с противоопухолевой активностью в Н2 клетки, стимулирующие пролиферацию опухолевых клеток, ангиогенез и метастазирования. Представлены данные о функционально важных цитокинах и хемокинах, выделяемых опухолевыми клетками, микроокружением, лимфоцитами и самими нейтрофилами, которые стимулируют выработку костным мозгом нейтрофилов и обусловливают накопление этих клеток в опухолевом очаге, развитие нейтрофилии в крови. Приведены основные функциональные молекулы нейтрофилов, такие как нейтрофильная эластаза, катепсин, матриксная металлопротеиназа-9, аргиназа 1 и др., с которыми связывают протуморогенные свойства Н2 нейтрофилов. Многие процессы, вызываемые в опухолевом очаге как Н1, так и Н2 нейтрофилами, еще не до конца изучены. Кратко отмечается о существовании сегодня многих подходов к генерации и активации нейтрофилов с противоопухолевыми свойствами и подавлению опухольстимулирующих нейтрофилов.
Приведены основные функциональные молекулы нейтрофилов, такие как нейтрофильная эластаза, катепсин, матриксная металлопротеиназа-9, аргиназа 1 и др., с которыми связывают протуморогенные свойства Н2 нейтрофилов. Многие процессы, вызываемые в опухолевом очаге как Н1, так и Н2 нейтрофилами, еще не до конца изучены. Кратко отмечается о существовании сегодня многих подходов к генерации и активации нейтрофилов с противоопухолевыми свойствами и подавлению опухольстимулирующих нейтрофилов.
Получено 09.02.2018
Принято в печать 16.03.2018
Введение
Общеизвестно, что нейтрофилы являются наиболее распространенными лейкоцитами крови и считаются первой линией защиты при воспалении и инфекциях [1]. Проникшие в организм микроорганизмы вызывают воспалительную реакцию, которая привлекает нейтрофилы из кровообращения в ткани. Там нейтрофилы разрушают микроорганизм с помощью ряда механизмов, главным образом за счет фагоцитоза, высвобождения противомикробных веществ и образования внеклеточных ловушек нейтрофилов [1, 2].
Помимо этой классической роли нейтрофилов в антимикробной защите, также выявлено накопление нейтрофилов во многих типах опухолей. Первоначально считалось, что эти связанные с опухолью нейтрофилы (опухольинфильтрирующие нейтрофилы — ОН) являются простыми свидетелями, потому что трудно представить, что нейтрофилы, будучи короткоживущими клетками, могут влиять на такое хроническое и прогрессирующее заболевание, как рак. Однако в последнее время стало известно, что ОН играют важную роль при злокачественных новообразованиях. Этот частично объясняется признанием того, что развитие воспаления, с одной стороны, в организме связано с нейтрофилами, а с другой, является важной характеристикой многих опухолей [6, 7].
Однако большее число клинических наблюдений и лабораторных исследований показали, что наличие нейтрофилов в опухолях часто коррелирует с плохим прогнозом. Это хорошо доказано при целом ряде опухолей, в частности при бронхоальвеолярной карциноме [10], почечно-клеточной карциноме [12] и плоскоклеточной карциноме головы и шеи [13], а также меланоме [11]. Во всех этих случаях нейтрофилы проявляют другой фенотип, который может быть неблагоприятным для исхода заболевания. Механизмы формирования и реализации этого фенотипа нейтрофилов только начинают выяснять, но предполагается, что некоторые из них связаны с генотоксичностью, ангиогенезом и иммуносупрессией [8].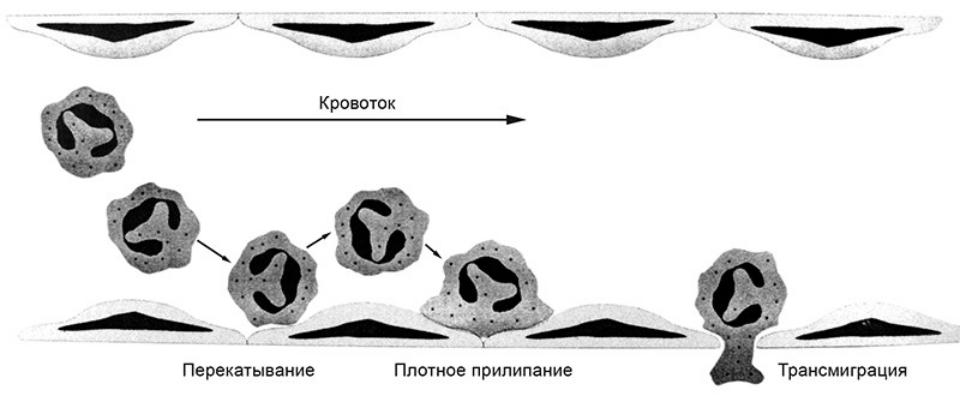
Стимуляция образования нейтрофилов. У многих пациентов с распространенным раком выявлены повышенные уровни нейтрофилов в крови. Как опухоли индуцируют нейтрофилию, окончательно неизвестно, но синтез опухолевыми клетками гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF), возможно, является одним из механизмов стимуляции продукции нейтрофилов костным мозгом [19]. Кроме того, и другие цитокины, такие как интерлейкин (IL)-1 и IL-6, продуцируемые также опухолями, по-видимому, способствуют увеличению числа нейтрофилов в крови [7, 20]. Эта нейтрофилия связана с плохим прогнозом при нескольких типах рака, таких как рак легкого, меланома и почечная карцинома [11, 21, 22]. В соответствии с этим наличие большого количества нейтрофилов в определенных типах опухолей также является показателем неблагоприятного прогноза. Поскольку нейтрофилия в крови часто ассоциируется с воспалительными реакциями на инфекцию и повреждение тканей, то в опухолевом очаге она представляет собой одно из доказательств концепции о роли воспаления в онкогенезе и индуцированного им прогрессирования роста опухоли [7].
В соответствии с этим наличие большого количества нейтрофилов в определенных типах опухолей также является показателем неблагоприятного прогноза. Поскольку нейтрофилия в крови часто ассоциируется с воспалительными реакциями на инфекцию и повреждение тканей, то в опухолевом очаге она представляет собой одно из доказательств концепции о роли воспаления в онкогенезе и индуцированного им прогрессирования роста опухоли [7].
Предполагается, что отношение числа нейтрофилов в крови к другим типам лейкоцитов служит фактором прогноза для больных раком. Так, например, отношение нейтрофилов к лимфоцитам (НЛО) было введено как прогностический фактор для больных колоректальным раком [25]. Из-за своей простоты определения НЛО показало, что является легкодоступным и недорогим биомаркером для многих типов опухолей, включая немелкоклеточный рак легкого [26], гепатоцеллюлярную карциному [24], карциному носоглотки [27], колоректальный рак [26], меланому [11] и рак грудной железы [28, 29]. Высокий уровень НЛО коррелирует с неблагоприятной общей выживаемостью при многих солидных опухолях [30–32].
В то же время, несмотря на данные многих исследований, часть которых упомянута выше, нейтрофилия (большее количество нейтрофилов в крови как следствие повышенного выхода клеток из костного мозга) не всегда является плохим показателем прогрессирования рака. При некоторых типах опухолей, например раке желудка, повышенный уровень нейтрофилов в крови сопряжен с положительным прогнозом [33]. Это означает, что нейтрофилы могут в некоторых случаях контролировать развитие рака. Способность нейтрофилов непосредственно убивать опухолевые клетки как in vitro, так и in vivo зарегистрирована давно [34–36]. Также сообщалось, что нейтрофилы от животных с экспериментальными опухолями обладают повышенной цитотоксической активностью [7]. Нейтрофилы, выделенные из крови здоровых людей, оказывают прямое цитотоксическое действие на некоторые линии опухолевых клеток [40]. Таким образом, точная роль нейтрофилов в развитии опухолевого роста различных типов рака является спорным вопросом [7, 14, 37] и не до конца изученной, что требует дальнейших исследований.
Типы нейтрофилов. В дополнение к увеличенному количеству нейтрофилов в крови отмечено повышение уровня в крови незрелых миелоидных клеток на ранних стадиях дифференцировки, что выявлено в нескольких типах опухолей [38], включая пациентов с терминальной стадией рака легкого, грудной железы и желудочно-кишечного тракта [39]. Эти незрелые клетки костномозгового происхождения, представляющие гетерогенную популяцию, фенотипически разделяли на гранулоцитарные (G-MDSC) и моноцитарные (Mo-MDSC) подгруппы [40–42]. Их выявляют в большом количестве в селезенке экспериментальных животных с опухолями, где они представляют иммунодепрессивный фенотип, что приводит к прогрессированию опухолей [43, 44]. G-MDSC характеризуются незрелой морфологией нейтрофилов и фенотипом CD33/CD11b/HLA-DR/CD15 у людей [45]. Они выявлены в периферической крови пациентов с глиобластомой [46], множественной миеломой, лимфомой Ходжкина [47], раком головы и шеи [48].
Эти G-MDSC могут осуществлять иммуносупрессию различными механизмами. Основной механизм включает в себя производство активных форм кислорода (АФК) при дыхательной вспышке этих клеток. У больных онкологического профиля пероксид водорода (H2O2), продуцируемый активированными гранулоцитами, снижал экспрессию CD3 цепи Т-клеточного рецептора и уменьшал выработку цитокинов Т-клетками пациентов [49]. Эти окисленные Т-клетки человека имели дефектный хемотаксис. Более того, АФК, продуцируемые MDSC, могут приводить к блокаде также CD8 T-клеток и с помощью другого механизма, в частности пероксинитрита [50].
Основной механизм включает в себя производство активных форм кислорода (АФК) при дыхательной вспышке этих клеток. У больных онкологического профиля пероксид водорода (H2O2), продуцируемый активированными гранулоцитами, снижал экспрессию CD3 цепи Т-клеточного рецептора и уменьшал выработку цитокинов Т-клетками пациентов [49]. Эти окисленные Т-клетки человека имели дефектный хемотаксис. Более того, АФК, продуцируемые MDSC, могут приводить к блокаде также CD8 T-клеток и с помощью другого механизма, в частности пероксинитрита [50].
Как отмечалось выше, в зависимости от фенотипа нейтрофилы можно классифицировать как Н1 или Н2 [15], и подобно инфильтрирующим опухоль макрофагам (M1) клетки Н1 проявляют провоспалительную и противоопухолевую функции. Напротив, клетки M2 и Н2 обладают протуморогенной активностью [16]. Установлено, что ОН отличаются от циркулирующих нейтрофилов, а также от G-MDSC в костном мозге и селезенке мышей. Мышиные CD11b/Ly6G нейтрофилы, выделенные из опухоли и активированные трансформирующим фактором роста бета (TGF-β), были гиперсегментированы и более цитотоксичны к опухолевым клеткам, экспрессировали более высокие уровни провоспалительных цитокинов [15].
Несмотря на эту классификацию ОН у мышей, природа и функция ОН, находящихся в опухолях человека, остаются еще малоизученными, но уже получены результаты, подтверждающие такое деление ОН. Так, в исследованиях биопсийного материала опухоли легкого человека ОН составляли 5–25% всех клеток в опухоли [65]. Эти ОН представляли активированный фенотип (CD62L/CD54) с экспрессией отчетливого репертуара рецепторов хемокинов, которые включали CCR5, CCR7, CXCR3 и CXCR4 [65]. Кроме того, ОН продуцировали большее количество провоспалительных факторов MCP-1, IL-8, MIP-1α и IL-6, чем нейтрофилы в крови. ОН также стимулировали пролиферацию Т-клеток и выделение интерферона-гамма (IFN-γ). Эти результаты показывают, что на ранних стадиях рака легкого ОН не являются иммунодепрессантами, а скорее всего стимулируют ответы Т-клеток [52]. В исследовании [53] изучена роль хронического воспаления, в частности IL-23 и IL-17, при раке толстой кишки человека. Авторы выявили, что врожденные γδT (γδT17) клетки являются основным клеточным источником IL-17 при колоректальном раке.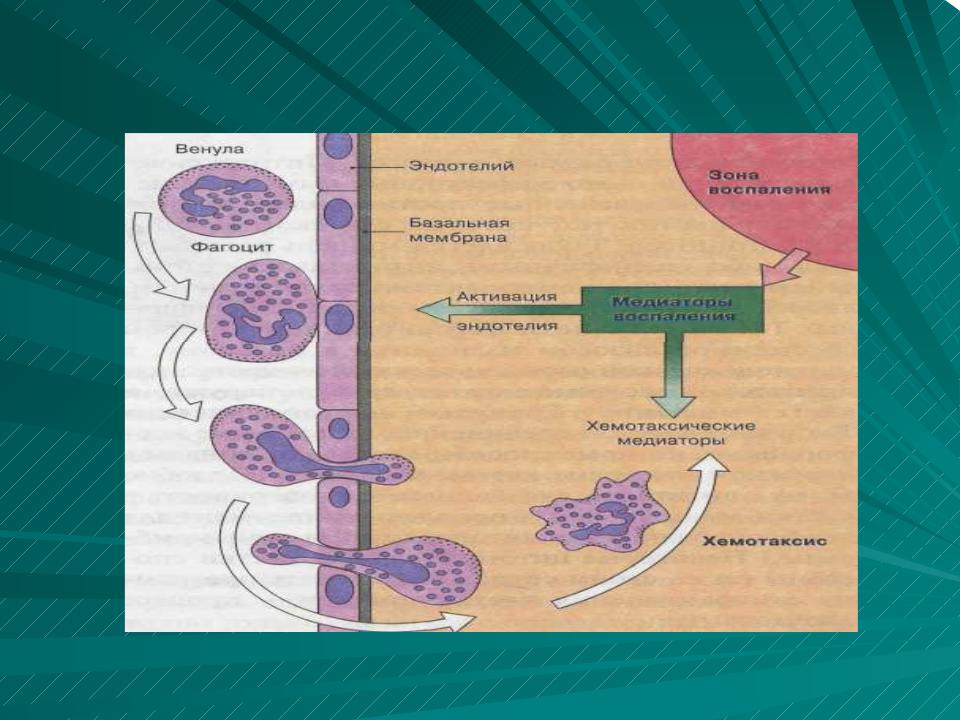 Эти активированные клетки индуцировали клетки γδT17 секретировать IL-8, TNF-α и GM-CSF, что приводит к накоплению нейтрофилов в опухоли. Эти ОН характеризовались CD45/Lin/HLADR/CD11b/CD33/CD66b и имели типичную полиморфноядерную морфологию. Они были описаны как G-MDSC [66]. Эти ОН (G-MDSC) продуцировали намного больше аргиназы-1 (ARG1) и АФК, чем аутологичные нейтрофилы в крови, и ингибировали пролиферацию активированных аутологичных Т-клеток и продукцию IFN-γ [53]. Результаты приведенных исследований указывают, что и в опухолях человека могут быть нейтрофилы с двойной функцией. В ранних стадиях развития опухоли ОН, по-видимому, способны стимулировать противоопухолевые иммунные реакции, особенно Т-клеток [52], на поздних этапах роста опухоли ОН уже становятся иммунодепрессивными клетками [54]. Сегодня остается еще много неясных вопросов. Например, являются ли ОН в ранних стадиях роста опухолей незрелыми нейтрофилами с противоопухолевыми свойствами, или ОН — это зрелые нейтрофилы, которые меняют фенотип со временем при прогрессировании опухоли, как предполагают ряд исследователей [17, 51].
Эти активированные клетки индуцировали клетки γδT17 секретировать IL-8, TNF-α и GM-CSF, что приводит к накоплению нейтрофилов в опухоли. Эти ОН характеризовались CD45/Lin/HLADR/CD11b/CD33/CD66b и имели типичную полиморфноядерную морфологию. Они были описаны как G-MDSC [66]. Эти ОН (G-MDSC) продуцировали намного больше аргиназы-1 (ARG1) и АФК, чем аутологичные нейтрофилы в крови, и ингибировали пролиферацию активированных аутологичных Т-клеток и продукцию IFN-γ [53]. Результаты приведенных исследований указывают, что и в опухолях человека могут быть нейтрофилы с двойной функцией. В ранних стадиях развития опухоли ОН, по-видимому, способны стимулировать противоопухолевые иммунные реакции, особенно Т-клеток [52], на поздних этапах роста опухоли ОН уже становятся иммунодепрессивными клетками [54]. Сегодня остается еще много неясных вопросов. Например, являются ли ОН в ранних стадиях роста опухолей незрелыми нейтрофилами с противоопухолевыми свойствами, или ОН — это зрелые нейтрофилы, которые меняют фенотип со временем при прогрессировании опухоли, как предполагают ряд исследователей [17, 51]. Уже идентифицировано несколько субпопуляций нейтрофилов в крови как мышей с опухолями, так и людей, больных раком, и описывается несколько вариантов взаимосвязей этих клеток в связи с прогрессированием рака [54]. Показано, что циркулирующие в крови нейтрофилы от животных с опухолями распределяли на субпопуляции (фракции) при разделении их в различных градиентах плотности фиколла. Одна субпопуляция состояла из «нормальных» нейтрофилов с высокой плотностью, которую выделяли на градиенте с высокой плотностью фиколла. Другая субпопуляция имела нейтрофилы с меньшей плотностью, которые находились вместе со слоем мононуклеарных лимфоидных клеток низкой плотности [55]. У здоровых мышей, не имеющих опухоли, большинство нейтрофилов были высокой плотности, а у животных с опухолями прогрессивно возрастало количество нейтрофилов с низкой плотностью, и они становились доминирующим типом нейтрофилов в циркуляции [54]. Эти нейтрофилы обладали меньшей цитотоксичностью и имели меньшую экспрессию различных хемокинов (CXCL1, CXCL2, CXCL10, CCL2 и CCL3) и хемокиновых рецепторов (CXCR2 и CCR5), что свидетельствовало о снижении их провоспалительных возможностей.
Уже идентифицировано несколько субпопуляций нейтрофилов в крови как мышей с опухолями, так и людей, больных раком, и описывается несколько вариантов взаимосвязей этих клеток в связи с прогрессированием рака [54]. Показано, что циркулирующие в крови нейтрофилы от животных с опухолями распределяли на субпопуляции (фракции) при разделении их в различных градиентах плотности фиколла. Одна субпопуляция состояла из «нормальных» нейтрофилов с высокой плотностью, которую выделяли на градиенте с высокой плотностью фиколла. Другая субпопуляция имела нейтрофилы с меньшей плотностью, которые находились вместе со слоем мононуклеарных лимфоидных клеток низкой плотности [55]. У здоровых мышей, не имеющих опухоли, большинство нейтрофилов были высокой плотности, а у животных с опухолями прогрессивно возрастало количество нейтрофилов с низкой плотностью, и они становились доминирующим типом нейтрофилов в циркуляции [54]. Эти нейтрофилы обладали меньшей цитотоксичностью и имели меньшую экспрессию различных хемокинов (CXCL1, CXCL2, CXCL10, CCL2 и CCL3) и хемокиновых рецепторов (CXCR2 и CCR5), что свидетельствовало о снижении их провоспалительных возможностей. Предполагается, что нейтрофилы с низкой плотность являются незрелыми нейтрофилами. Важно отметить, что TGF-β способен индуцировать трансформацию нейтрофилов с высокой плотностью от мышей с опухолями в нейтрофилы с низкой плотностью, и в то же время этот фактор не влиял на нейтрофилы крови животных без опухолей [54]. Это указывает на то, что для такого изменения ОН у животных с раком необходимы и другие стимулы, помимо TGF-β. На основании этих результатов авторы предложили гипотезу, согласно которой три субпопуляции нейтрофилов могут присутствовать в крови при раке: нормальные нейтрофилы высокой плотности, незрелые нейтрофилы низкой плотности (G-MDSC) и крупные зрелые нейтрофилы низкой плотности. Эти типы клеток обладают разной функциональностью и пластичностью. Так, нейтрофилы с высокой плотностью являются противоопухолевыми, а низкой — клетками, способными стимулировать рост опухоли [67], они могут изменяться под влиянием различных хемокинов и цитокинов в микроокружении опухоли [17].
Предполагается, что нейтрофилы с низкой плотность являются незрелыми нейтрофилами. Важно отметить, что TGF-β способен индуцировать трансформацию нейтрофилов с высокой плотностью от мышей с опухолями в нейтрофилы с низкой плотностью, и в то же время этот фактор не влиял на нейтрофилы крови животных без опухолей [54]. Это указывает на то, что для такого изменения ОН у животных с раком необходимы и другие стимулы, помимо TGF-β. На основании этих результатов авторы предложили гипотезу, согласно которой три субпопуляции нейтрофилов могут присутствовать в крови при раке: нормальные нейтрофилы высокой плотности, незрелые нейтрофилы низкой плотности (G-MDSC) и крупные зрелые нейтрофилы низкой плотности. Эти типы клеток обладают разной функциональностью и пластичностью. Так, нейтрофилы с высокой плотностью являются противоопухолевыми, а низкой — клетками, способными стимулировать рост опухоли [67], они могут изменяться под влиянием различных хемокинов и цитокинов в микроокружении опухоли [17].
Очевидно, высокий уровень нейтрофилов в опухоли происходит под действием нейтрофил-притягивающих хемокинов, которые могут продуцироваться не только иммунными клетками, но и опухолевыми клетками, в частности интерлейкином-8 (IL-8/CXCL8). Другим хемокином, который также участвует в рекрутировании нейтрофилов в опухоли, является хемокин GCP-2/CXCL6. В мышиной модели меланомы специфические моноклональные антитела против CXCL6 уменьшали количество ОН, а также размер опухоли [56]. Кроме того, из карциноматозных опухолей человека выделен и идентифицирован фактор ингибирования миграции (MIF), уровень которого был высокий в опухолях с бóльшим содержанием ОН и низкой выживаемостью этих пациентов [57]. Используя те же рецепторы CXCR1 и CXCR2, нейтрофилы могут также реагировать и на другие хемокины, такие как CXCL1, CXCL2, CXCL5, CXCL6 и CXCL7 [58]. Не исключено, что нейтрофилы активируют сами себя по механизму положительной обратной связи, высвобождая нейтрофильные хемокины, которые привлекают больше нейтрофилов в опухоль, подобно миграции нейтрофилов в очаги инфекции [59]. При исследовании 919 пациентов с гепатоцеллюлярной карциномой выявлено, что CXCL5 сверхэкспрессируется у пациентов с рецидивирующими опухолями, уровни CXCL5 коррелировали с большим накоплением в опухоли нейтрофилов и с меньшей общей выживаемостью [60].
Другим хемокином, который также участвует в рекрутировании нейтрофилов в опухоли, является хемокин GCP-2/CXCL6. В мышиной модели меланомы специфические моноклональные антитела против CXCL6 уменьшали количество ОН, а также размер опухоли [56]. Кроме того, из карциноматозных опухолей человека выделен и идентифицирован фактор ингибирования миграции (MIF), уровень которого был высокий в опухолях с бóльшим содержанием ОН и низкой выживаемостью этих пациентов [57]. Используя те же рецепторы CXCR1 и CXCR2, нейтрофилы могут также реагировать и на другие хемокины, такие как CXCL1, CXCL2, CXCL5, CXCL6 и CXCL7 [58]. Не исключено, что нейтрофилы активируют сами себя по механизму положительной обратной связи, высвобождая нейтрофильные хемокины, которые привлекают больше нейтрофилов в опухоль, подобно миграции нейтрофилов в очаги инфекции [59]. При исследовании 919 пациентов с гепатоцеллюлярной карциномой выявлено, что CXCL5 сверхэкспрессируется у пациентов с рецидивирующими опухолями, уровни CXCL5 коррелировали с большим накоплением в опухоли нейтрофилов и с меньшей общей выживаемостью [60]. В частности, известно, что активированные Т-клетки продуцируют GM-CSF, CXCL1, CXCL2, TNF-α и IFN-γ [59]. Эти факторы могут прямо или косвенно привлекать больше нейтрофилов к опухоли. Хотя конкретные механизмы влияния активированных Т-клеток на миграцию нейтрофилов в опухоль изучены недостаточно.
В частности, известно, что активированные Т-клетки продуцируют GM-CSF, CXCL1, CXCL2, TNF-α и IFN-γ [59]. Эти факторы могут прямо или косвенно привлекать больше нейтрофилов к опухоли. Хотя конкретные механизмы влияния активированных Т-клеток на миграцию нейтрофилов в опухоль изучены недостаточно.
Нейтрофильные молекулы. Большой объем клинических данных показывает, что нейтрофилы участвуют в развитии рака и прогрессировании опухолей. В большинстве случаев увеличенное количество ОН ассоциируется с прогрессирующей болезнью и плохим прогнозом для больных онкологического профиля. Установлено, что такая отрицательная ассоциация характерна для ряда солидных опухолей, таких как меланома, гепатоцеллюлярная карцинома, немелкоклеточная карцинома легкого, глиома, аденокарцинома и рак толстой кишки [37, 61]. Предполагается, что в стимуляцию онкогенеза включаются те же молекулы, которые нейтрофилы используют для уничтожения микроорганизмов и модуляции воспаления [7]. Важными молекулами, которые могут влиять на темп роста и инвазивность опухолей, являются гранулярные белки, деградирующие в матриксе протеиназы, реактивные виды кислорода, хемокины и цитокины [7]. В последнее время появились сообщения, описывающие, как ОН используют эти молекулы для воздействия на пролиферацию опухолевых клеток, ангиогенез, метастазы и иммунный надзор. Среди этих молекул следует выделить такие, как нейтрофильная эластаза (NE), катепсин, матриксная металлопротеиназа (MMP)-9 и др.
В последнее время появились сообщения, описывающие, как ОН используют эти молекулы для воздействия на пролиферацию опухолевых клеток, ангиогенез, метастазы и иммунный надзор. Среди этих молекул следует выделить такие, как нейтрофильная эластаза (NE), катепсин, матриксная металлопротеиназа (MMP)-9 и др.
NE представляет собой основной белок азурофильных гранул, который выделяется при клеточной дегрануляции нейтрофилов. NE — сериновая протеаза с широким спектром субстратов. Помимо своей роли в воспалении и уничтожении бактерий, NE проявляет различные протуморогенные эффекты как in vivo, так и in vitro [62]. Выявлено, что NE непосредственно стимулирует пролиферацию опухолевых клеток A459, когда мышиные нейтрофилы культивировали вместе с клеточной линией карциномы легкого [62, 63]. Кроме того, также установлено, что NE стимулирует миграцию опухолевых клеток. Нейтрофилы человека при культивировании с клетками аденокарциномы поджелудочной железы стимулировали миграцию клеток опухоли из монослоя. NE также повышала миграционную способность раковых клеток пищевода [64].
NE также повышала миграционную способность раковых клеток пищевода [64].
Катепсин G представляет собой пептидазу из азурофильных гранул, которая участвует в деградации фагоцитированных микроорганизмов и ремоделировании белков внеклеточного матрикса [98]. Кроме того, катепсин G может стимулировать ангиогенез и миграцию опухолевых клеток [65–67]. На модели метастазирования рака грудной железы в костную ткань также показано, что катепсин G усиливает передачу сигналов TGF-β и повышает уровень сосудистого эндотелиального фактора роста (VEGF) для стимуляции ангиогенеза [66].
MMP-9 — желатиназа B — высвобождается из вторичных (специфических) гранул и, как установлено, приводит к пролиферации опухолей кожи человека вирусом папилломы человека 16-го типа (HPV-16). Кроме того, иммуногистохимическое исследование MMP-9 в плоскоклеточных опухолях карциномы показало, что MMP-9 присутствовала только в опухолевых инфильтрирующих лейкоцитах, а не в самих опухолевых клетках. Показано, что MMP-9 ингибирует апоптоз опухолевых клеток при раке легкого [68]. Таким образом, ММП-9 ответственна за усиление роста опухоли как за счет увеличения пролиферации клеток, так и уменьшения их апоптоза. Другим важным эффектом MMP-9, который поддерживает рост опухоли, является ангиогенез. Протеолитическое высвобождение VEGF из тканевого матрикса при действии MMPs считается необходимым условием ангиогенеза in vivo [68, 69]. Ангиогенный эффект MMP-9 зарегистрирован в нескольких моделях рака — меланоме, аденокарциноме поджелудочной железы [70–72].
Таким образом, ММП-9 ответственна за усиление роста опухоли как за счет увеличения пролиферации клеток, так и уменьшения их апоптоза. Другим важным эффектом MMP-9, который поддерживает рост опухоли, является ангиогенез. Протеолитическое высвобождение VEGF из тканевого матрикса при действии MMPs считается необходимым условием ангиогенеза in vivo [68, 69]. Ангиогенный эффект MMP-9 зарегистрирован в нескольких моделях рака — меланоме, аденокарциноме поджелудочной железы [70–72].
Прямое доказательство того, что нейтрофилы являются основным, ассоциированным с опухолью лейкоцитарным типом, экспрессирующим MMP-9, предоставлено в исследовании с использованием человеческих ксенотрансплантатов и сингенных опухолей в эксперименте на мышах [73]. Когда опухоли или изолированные из них макрофаги или нейтрофилы были дважды окрашены для выявления MMP-9 и соответствующих антигенов макрофагов или нейтрофилов, только ОН содержали большое количество MMP-9 [74, 75]. Кроме того, подсчитано, что 1•10 нейтрофилов в крови или ОН могут высвобождать приблизительно 100–200 нг проМMP-9 в течение 1–2 ч инкубации. Напротив, для макрофагов 1•10 потребуется несколько недель для получения такого же количества проММР-9 [73, 74]. Следовательно, MMP-9, полученная из нейтрофилов, ответственна за усиление ангиогенеза за счет высвобождения VEGF из внеклеточного матрикса, что отмечается при многих типах опухолей. С внеклеточным матриксом связывают и другие процессы онкогенеза, например метастазирование опухолей [75, 76].
Напротив, для макрофагов 1•10 потребуется несколько недель для получения такого же количества проММР-9 [73, 74]. Следовательно, MMP-9, полученная из нейтрофилов, ответственна за усиление ангиогенеза за счет высвобождения VEGF из внеклеточного матрикса, что отмечается при многих типах опухолей. С внеклеточным матриксом связывают и другие процессы онкогенеза, например метастазирование опухолей [75, 76].
Нейтрофилы являются эффективными производителями АФК для уничтожения микроорганизмов. АФК также может косвенно способствовать росту опухоли. Во-первых, нейтрофилы генерируют пероксид водорода (H2O2), который затем превращается в гипохлорид (HOCl) с помощью миелопероксидазы. HOCl затем может активировать несколько MMPs, включая MMP-2, -7, -8 и -9. Кроме того, HOCl может блокировать ингибитор МMP-1 и таким образом потенцировать протеолитическую активность MMPs [77, 78].
Выделенная из гранул нейтрофилов ARG1 способна разрушать внеклеточный аргинин, незаменимую аминокислоту для активации Т-клеток. Таким образом, дегрануляция нейтрофилов может оказывать иммуносупрессивный эффект в опухолях, ингибируя опухольинфильтрирующие Т-клетки таким же образом, как описано для G-MDSC [79]. Показано, что истощение ОН у опухольсодержащих животных увеличило число активированных CD8 T-клеток, способствовало уменьшению размера опухолей и удлиняло время жизни животных [15].
Таким образом, дегрануляция нейтрофилов может оказывать иммуносупрессивный эффект в опухолях, ингибируя опухольинфильтрирующие Т-клетки таким же образом, как описано для G-MDSC [79]. Показано, что истощение ОН у опухольсодержащих животных увеличило число активированных CD8 T-клеток, способствовало уменьшению размера опухолей и удлиняло время жизни животных [15].
Нейтрофилы могут также продуцировать цитокины или факторы роста, которые увеличивают туморогенный потенциал раковых клеток [5]. Это пока что установлено для двух цитокинов — онкостатина-M [79–81] и для фактора роста гепатоцитов [10, 82, 83]. Раковые клетки грудной железы могут стимулировать нейтрофилы к выделению онкостатина-М, IL-6-подобного цитокина. Онкостатин-М, в свою очередь, стимулировал клетки рака грудной железы секретировать VEGF [84]. Аналогично, клетки гепатоцеллюлярной карциномы стимулировали нейтрофилы высвобождать фактор роста гепатоцитов (HGF). В свою очередь, этот фактор стимулировал инвазивный рост опухолевых клеток [85].
Нейтрофилы также могут влиять на миграционный потенциал раковых клеток. При нескольких типах рака показано, что нейтрофилы способствуют метастазированию плоскоклеточного рака кожи [86], меланомы [87], аденокарциномы [88] и рака грудной железы [89]. Способ, которым нейтрофилы повышают миграционную активность опухолевых клеток, может включать несколько различных механизмов. Циркулирующие опухолевые клетки непосредственно прилипают к сосудистому эндотелию, приводя к экстравазации для создания новых метастазов. В месте образования метастатического очага клетками рака легкого отмечалась их тесная связь с нейтрофилами [90]. В этом процессе нейтрофилы усиливают задержку опухолевых клеток, и, как следствие, возникает больше метастазов [91]. Показано прямое взаимодействие между клетками нейтрофилов и клетками карциномы грудной железы путем взаимодействия молекул адгезии ICAM-1 в опухолевых клетках и β2-интегринов на нейтрофилах. Нейтрофилы связывали опухолевые клетки с участием интегринов и индуцировали кластеризацию ICAM-1 в опухолевой клетке [91]. Это активировало в опухолевой клетке сигнальный путь с участием фокальной адгезии киназы (FAK) и p38-MAPK, что привело к усиленной миграции [89]. Вследствие этого повышенная миграция, как показано in vivo, приводила к увеличению количества метастазов в печени [92]. Раковые клетки непосредственно прикреплялись поверх адгезированных нейтрофилов, которые выступали в качестве мостика при взаимодействии между опухолевыми клетками и паренхимой печени, что ускоряло развитие метастазов [93].
Это активировало в опухолевой клетке сигнальный путь с участием фокальной адгезии киназы (FAK) и p38-MAPK, что привело к усиленной миграции [89]. Вследствие этого повышенная миграция, как показано in vivo, приводила к увеличению количества метастазов в печени [92]. Раковые клетки непосредственно прикреплялись поверх адгезированных нейтрофилов, которые выступали в качестве мостика при взаимодействии между опухолевыми клетками и паренхимой печени, что ускоряло развитие метастазов [93].
Несмотря на большое количество доказательств отрицательной роли нейтрофилов во время прогрессирования опухоли, имеются также четкие свидетельства положительной роли нейтрофилов в канцерогенезе. Как упоминалось ранее, нейтрофилы могут проявлять противоопухолевую активность в различных формах. Фактически, противоопухолевая способность нейтрофилов установлена давно, более трех десятилетий назад. Нейтрофилы могут непосредственно уничтожать опухолевые клетки как in vitro [36], так и in vivo [37]. Нейтрофилы потенцируют этот противоопухолевый эффект, когда они активированы. Важность ОН типа Н1 в противоопухолевых реакциях также подчеркивается экспериментальными исследованиями о том, что истощение уровня нейтрофилов в крови приводит к увеличению роста опухоли [15, 93, 94]. Очевидно, что нейтрофилы обладают потенциалом непосредственного уничтожения опухолевых клеток. Механизмы, посредством которых нейтрофилы выполняют эту функцию, многочисленны и еще не полностью изучены, но они включают в себя многие уже известные механизмы антиинфекционной защиты [7].
Нейтрофилы потенцируют этот противоопухолевый эффект, когда они активированы. Важность ОН типа Н1 в противоопухолевых реакциях также подчеркивается экспериментальными исследованиями о том, что истощение уровня нейтрофилов в крови приводит к увеличению роста опухоли [15, 93, 94]. Очевидно, что нейтрофилы обладают потенциалом непосредственного уничтожения опухолевых клеток. Механизмы, посредством которых нейтрофилы выполняют эту функцию, многочисленны и еще не полностью изучены, но они включают в себя многие уже известные механизмы антиинфекционной защиты [7].
ЗАКЛЮЧЕНИЕ
Таким образом, согласно представленным выше кратким сведениям подчеркивается двойной противоопухолевый и протуморальный потенциал нейтрофилов и предполагается, что нейтрофилы могут быть использованы для усиления различных противоопухолевых реакций в организме.
Хотя во многих случаях наличие нейтрофилов в опухолях оказывает негативное влияние на течение раковой болезни, эти клетки, несомненно, обладают способностью разрушать опухолевые клетки. Сегодня рассматривается около десяти новых терапевтических стратегий и подходов для усиления противоопухолевого потенциала нейтрофилов или блокирования доступа ОН к растущим опухолям, такие как активация нейтрофилов интерфероном, синтез и накопление высоких уровней провоспалительных цитокинов в опухоли, которые могут убивать опухолевые клетки, блокирование инфильтрации нейтрофилами опухоли и многие другие. Как указывалось выше, в некоторых опухолях образуются хемокины, главным образом IL-8, которые привлекают нейтрофилы в опухолевый очаг. Показано, что применение антагонистов IL-8 (таких как полностью гуманизированное нейтрализующее моноклональное антитело ABX-IL8) к IL-8 уменьшает рост опухоли, метастазы и ангиогенез меланомы и рака легкого [95]. Наличие двух фенотипов Н1 и Н2 нейтрофилов, предполагает также, что, влияя на микроокружение опухоли, можно манипулировать ОН и генерировать большее количество противоопухолевых нейтрофилов. Также получены многообещающие результаты с использованием моноклональных терапевтических антител, индуцирующих нейтрофилы для выполнения антителозависимой цитотоксичности и высвобождения цитокинов, которые модулируют иммунный противоопухолевый ответ [96, 97].
Сегодня рассматривается около десяти новых терапевтических стратегий и подходов для усиления противоопухолевого потенциала нейтрофилов или блокирования доступа ОН к растущим опухолям, такие как активация нейтрофилов интерфероном, синтез и накопление высоких уровней провоспалительных цитокинов в опухоли, которые могут убивать опухолевые клетки, блокирование инфильтрации нейтрофилами опухоли и многие другие. Как указывалось выше, в некоторых опухолях образуются хемокины, главным образом IL-8, которые привлекают нейтрофилы в опухолевый очаг. Показано, что применение антагонистов IL-8 (таких как полностью гуманизированное нейтрализующее моноклональное антитело ABX-IL8) к IL-8 уменьшает рост опухоли, метастазы и ангиогенез меланомы и рака легкого [95]. Наличие двух фенотипов Н1 и Н2 нейтрофилов, предполагает также, что, влияя на микроокружение опухоли, можно манипулировать ОН и генерировать большее количество противоопухолевых нейтрофилов. Также получены многообещающие результаты с использованием моноклональных терапевтических антител, индуцирующих нейтрофилы для выполнения антителозависимой цитотоксичности и высвобождения цитокинов, которые модулируют иммунный противоопухолевый ответ [96, 97].
На развитие опухоли оказывают влияние многие типы клеток организма, в том числе ОН. Точная роль ОН до конца не установлена, и ее предстоит еще выяснить. В настоящее время широко изучаются разные способы привлечения их в опухоль и превращения Н1 нейтрофилов в противоопухолевые эффекторные клетки. Научиться переворачивать «монеты» нейтрофилов на «выигрышную сторону», как считают E. Uribe-Querol и C. Rosales [98], а именно, заставить действовать их как противоопухолевые эффекторные клетки, является вызовом и задачей для будущих исследований, что, возможно, позволит усовершенствовать существующие методы лечения рака.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Borregaard N. (2010) Neutrophils, from marrow to microbes. Immunity, 33(5): 657–670. doi: 10.1016/j.immuni.2010.11.011.
2. Kolaczkowska E., Kubes P. (2013) Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol., 13(3): 159–175. doi: 10.1038/nri3399.
3. Pham C.T.N. (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol., 6(7): 541–550. doi: 10.1038/nri1841.
Nat. Rev. Immunol., 6(7): 541–550. doi: 10.1038/nri1841.
4. Scapini P., Lapinet-Vera J.A., Gasperini S. et al. (2000) The neutrophil as a cellular source of chemokines. Immunol. Rev., 177: 195–203. doi: 10.1034/j.1600-065x.2000.17706.
5. Tecchio C., Scapini P., Pizzolo G., Cassatella M.A. (2013) On the cytokines produced by human neutrophils in tumors. Sem. Cancer Biol., 23(3): 159–170. doi: 10.1016/j.semcancer.2013.02.004.
6. Mantovani A., Allavena P., Sica A., Balkwill F. (2008) Cancer-related inflammation. Nature, 454 (7203): 436–444. doi: 10.1038/nature07205.
7. Бережная Н.М., Чехун В.Ф. (2005) Иммунология злокачественного роста. Наукова думка, Киев, 791 с.
8. Gregory A.D., Houghton A.M. (2011) Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res., 71(7): 2411–2416. doi: 10.1158/0008-5472.can-10-2583.
9. Mantovani A., Cassatella M.A., Costantini C., Jaillon S. (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol., 11(8): 519–531. doi: 10.1038/nri3024.
Nat. Rev. Immunol., 11(8): 519–531. doi: 10.1038/nri3024.
10. Wislez M., Rabbe N., Marchal J. et al. (2003) Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res., 63(6): 1405–1412.
11. Schmidt H., Bastholt L., Geertsen P. et al. (2005) Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: a prognostic model. Brit. J. Cancer, 93(3): 273–278. doi: 10.1038/sj.bjc.6602702.
12. Jensen H.K., Donskov F., Marcussen N. et al. (2009) Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J. Clin. Oncol., 27(28): 4709–4717. doi: 10.1200/jco.2008.18.9498.
13. Trellakis S., Bruderek K., Dumitru C.A. et al. (2011) Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int. J. Cancer, 129(9): 2183–2193. doi: 10.1002/ijc.25892.
Int. J. Cancer, 129(9): 2183–2193. doi: 10.1002/ijc.25892.
14. Fridlender Z.G., Albelda S.M. (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis, 33(5): 949–955. doi: 10.1093/carcin/bgs123.
15. Fridlender Z.G., Sun J., Kim S. et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-β: ‘N1’ versus ‘N2’ TAN. Cancer Cell, 16(3): 183–194. doi: 10.1016/j.ccr.2009.06.017. [PMC free article].
16. Galdiero M.R., Garlanda C., Jaillon S. et al. (2013) Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol., 228(7): 1404–1412. doi: 10.1002/jcp.24260.
17. Sionov R.V., Fridlender Z.G., Granot Z. (2014) The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron., 1–34. doi: 10.1007/s12307-014-0147-5.
18. Swierczak A., Mouchemore K.A., Hamilton J.A., Anderson R.L. (2015) Neutrophils: important contributors to tumor progression and metastasis. Cancer and Metastasis Rev., 34(4): 735–751. doi: 10.1007/s10555-015-9594-9.
doi: 10.1007/s10555-015-9594-9.
19. McGary C.T., Miele M.E., Welch D.R. (1995) Highly metastatic 13762NF rat mammary adenocarcinoma cell clones stimulate bone marrow by secretion of granulocyte-macrophage colony-stimulating factor/interleukin-3 activity. Am. J. Pathol., 147(6): 1668–1681.
20. Lechner M.G., Liebertz D.J., Epstein A.L. (2010) Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol., 185(4): 2273–2284. doi: 10.4049/jimmunol.1000901.
21. Atzpodien J., Reitz M. (2008) Peripheral blood neutrophils as independent immunologic predictor of response and long-term survival upon immunotherapy in metastatic renal-cell carcinoma. Cancer Biother. Radiopharm., 23(1): 129–134. doi: 10.1089/cbr.2007.0429.
22. Bellocq A., Antoine M., Flahault A. et al. (1998) Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am. J. Pathol., 152(1): 83–92.
Am. J. Pathol., 152(1): 83–92.
23. Reid M.D., Basturk O., Thirabanjasak D. et al. (2011) Tumor-infiltrating neutrophils in pancreatic neoplasia. Modern Pathol., 24(12): 1612–1619. doi: 10.1038/modpathol.2011.113.
24. Halazun K.J., Hardy M.A., Rana A.A. et al. (2009) Negative impact of neutrophil-lymphocyte ratio on outcome after liver transplantation for hepatocellular carcinoma. Ann. Surg., 250(1): 141–151. doi: 10.1097/SLA.0b013e3181a77e59.
25. Walsh S.R., Cook E.J., Goulder F. et al. (2005) Neutrophil-lymphocyte ratio as a prognostic factor in colorectal cancer. J. Surg. Oncol., 91(3): 181–184. doi: 10.1002/jso.20329.
26. Peng B., Wang Y.-H., Liu Y.-M., Ma L.-X. (2015) Prognostic significance of the neutrophil to lymphocyte ratio in patients with non-small cell lung cancer: a systemic review and meta-analysis. Int. J. Clin. Exp. Med., 8(3): 3098–3106.
27. Malietzis G., Giacometti M., Kennedy R.H. et al. (2014) The emerging role of neutrophil to lymphocyte ratio in determining colorectal cancer treatment outcomes: a systematic review and meta-analysis. Ann. Surg. Oncol., 21(12): 3938–3946. doi: 10.1245/s10434-014-3815-2.
Ann. Surg. Oncol., 21(12): 3938–3946. doi: 10.1245/s10434-014-3815-2.
28. Krenn-Pilko S., Langsenlehner U., Thurner E.-M. et al. (2014) The elevated preoperative derived neutrophil-to-lymphocyte ratio predicts poor clinical outcome in breast cancer patients. Brit. J. Cancer, 110(10): 2524–2530. doi: 10.1038/bjc.2014.163.
29. Pistelli M., De Lisa M., Ballatore Z. et al. (2015) Pre-treatment neutrophil to lymphocyte ratio may be a useful tool in predicting survival in early triple negative breast cancer patients. BMC Cancer, 15, article 195. doi: 10.1186/s12885-015-1204-2.
30. Guthrie G.J.K., Charles K.A., Roxburgh C.S.D. et al. (2013) The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit. Rev. Oncol. Hematol., 88(1): 218–230. doi: 10.1016/j.critrevonc.2013.03.010.
31. Templeton A.J., McNamara M.G., Šeruga B. et al. (2014) Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Nat. Cancer Institute, 106(6). doi: 10.1093/jnci/dju124.dju124.
J. Nat. Cancer Institute, 106(6). doi: 10.1093/jnci/dju124.dju124.
32. Paramanathan A., Saxena A., Morris D.L. (2014) A systematic review and meta-analysis on the impact of pre-operative neutrophil lymphocyte ratio on long term outcomes after curative intent resection of solid tumours. Surg. Oncol., 23(1): 31–39. doi: 10.1016/j.suronc.2013.12.001.
33. Caruso R.A., Bellocco R., Pagano M. et al. (2002) Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in Northern Italy. Modern Pathol., 15(8): 831–837. doi: 10.1097/01.mp.0000020391.98998.6b.
34. Pickaver A.H., Ratcliffe N.A., Williams A.E., Smith H. (1972) Cytotoxic effects of peritoneal neutrophils on a syngeneic rat tumour. Nature: New biology, 235(58): 186–187.
35. Gerrard T.L., Cohen D.J., Kaplan A.M. (1981) Human neutrophil-mediated cytotoxicity to tumor cells. J. Natl Cancer Inst., 66(3): 483–488 (19820701) 50:160;62::aid-cncr282050011362;3.0.co;2-0.
36. Katano M., Torisu M. (1982) Neutrophil-mediated tumor cell destruction in cancer ascites. Cancer, 50(1): 62–68. doi: 10.1002/1097-0142.
Katano M., Torisu M. (1982) Neutrophil-mediated tumor cell destruction in cancer ascites. Cancer, 50(1): 62–68. doi: 10.1002/1097-0142.
37. Brandau S., Dumitru C.A., Lang S. (2013) Protumor and antitumor functions of neutrophil granulocytes. Seminars in Immunopathology, 35(2): 163–176. doi: 10.1007/s00281-012-0344-6.
38. Almand B., Clark J.I., Nikitina E. et al. (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol., 166(1): 678–689. doi: 10.4049/jimmunol.166.1.678.
39. Choi J., Suh B., Ahn Y. et al. (2012) CD15+/CD16 human granulocytes from terminal cancer patients: granulocytic myeloid-derived suppressor cells that have suppressive function. Tumor Biology, 33(1): 121–129. doi: 10.1007/s13277-011-0254-6.
40. Peranzoni E., Zilio S., Marigo I. et al. (2010) Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol., 22(2): 238–244. doi: 10.1016/j.coi.2010. 01.021.
01.021.
41. Raber P.L., Thevenot P., Sierra R. et al. (2014) Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer, 134(12): 2853–2864. doi: 10.1002/ijc.28622.
42. Youn J.-I., Nagaraj S., Collazo M., Gabrilovich D.I. (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol., 181(8): 5791–5802. doi: 10.4049/jimmunol.181.8.5791.
43. Gabrilovich D.I., Nagaraj S. (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol., 9(3): 162–174. doi: 10.1038/nri2506.
44. Nagaraj S., Schrum A.G., Cho H.-I. et al. (2010) Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol., 184(6): 3106–3116. doi: 10.4049/jimmunol.0902661.
45. Favaloro J., Liyadipitiya T., Brown R. et al. (2014) Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leukemia & Lymphoma, 55(12): 2893–2900. doi: 10.3109/10428194.2014.904511.
Leukemia & Lymphoma, 55(12): 2893–2900. doi: 10.3109/10428194.2014.904511.
46. Raychaudhuri B., Rayman P., Huang P. et al. (2015) Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neuro-Oncology, 122: 293–301. doi: 10.1007/s11060-015-1720-6.
47. Gallamini A., Di Raimondo F., La Nasa G. et al. (2013) Standard therapies versus novel therapies in Hodgkin lymphoma. Immunol. Letters, 155(1–2): 56–59. doi: 10.1016/j.imlet.2013.09.011.
48. Trellakis S., Bruderek K., Hütte J. et al. (2013) Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer. Innate Immunity, 19(3): 328–336. doi: 10.1177/1753425912463618.
49. Schmielau J., Finn O.J. (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res., 61(12): 4756–4760.
Cancer Res., 61(12): 4756–4760.
50. Nagaraj S., Gupta K., Pisarev V. et al. (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med., 13(7): 828–835. doi: 10.1038/nm1609.
51. Mishalian I., Bayuh R., Levy L. et al. (2013) Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol., Immunother., 62(11): 1745–1756. doi: 10.1007/s00262-013-1476-9.
52. Eruslanov E.B., Bhojnagarwala P.S., Quatromoni J.G. et al. (2014) Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Invest., 124(12): 5466–5480. doi: 10.1172/JCI77053.
53. Wu P., Wu D., Ni C. et al. (2014) γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity, 40(5): 785–800. doi: 10.1016/j.immuni.2014.03.013.
54. Sagiv J.Y., Michaeli J., Assi S. et al. (2015) Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer.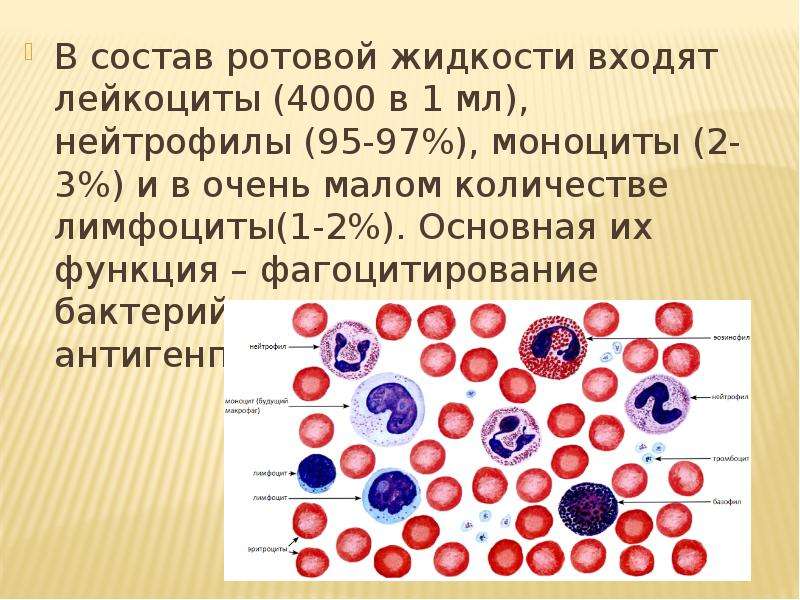 Cell Reports, 10(4): 562–573. doi: 10.1016/j.celrep.2014.12.039.
Cell Reports, 10(4): 562–573. doi: 10.1016/j.celrep.2014.12.039.
55. García-García E., Uribe-Querol E., Rosales C. (2013) A simple and efficient method to detect nuclear factor activation in human neutrophils by flow cytometry. J. Vis. Exp. (74). doi: 10.3791/50410.e50410 [PMC free article] [PubMed] [Cross Ref10.1016/j.celrep.2014.12.039].
56. Verbeke H., Struyf S., Berghmans N. et al. (2011) Isotypic neutralizing antibodies against mouse GCP-2/CXCL6 inhibit melanoma growth and metastasis. Cancer Letters, 302(1): 54–62. doi: 10.1016/j.canlet.2010.12.013.
57. Dumitru C.A., Gholaman H., Trellakis S. et al. (2011) Tumor-derived macrophage migration inhibitory factor modulates the biology of head and neck cancer cells via neutrophil activation. Int. J. Cancer, 129(4): 859–869. doi: 10.1002/ijc.25991. [PubMed] [Cross Ref.].
58. Lazennec G., Richmond A. (2010) Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med., 16(3): 133–144. doi: 10.1016/j.molmed.2010.01.003. [PMC free article] [PubMed] [Cross Ref.].
doi: 10.1016/j.molmed.2010.01.003. [PMC free article] [PubMed] [Cross Ref.].
59. Kobayashi Y. (2008) The role of chemokines in neutrophil biology. Front. Bioscie., 13(7): 2400–2407. doi: 10.2741/2853. [PubMed] [Cross Ref.].
60. Okabe H., Beppu T., Ueda M. et al. (2012) Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int. J. Cancer, 131(10): 2234–2241. doi: 10.1002/ijc.27496.
61. Dumitru C.A., Moses K., Trellakis S. et al. (2012) Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol., Immunother., 61(8): 1155–1167. doi: 10.1007/s00262-012-1294-5.
62. Houghton A.M., Rzymkiewicz D.M., Ji H. et al. (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med., 16(2): 219–223. doi: 10.1038/nm.2084.
63. Wada Y., Yoshida K., Tsutani Y. et al. (2007) Neutrophil elastase induces cell proliferation and migration by the release of TGF-α, PDGF and VEGF in esophageal cell lines. Oncol. Reports, 17(1): 161–167.
(2007) Neutrophil elastase induces cell proliferation and migration by the release of TGF-α, PDGF and VEGF in esophageal cell lines. Oncol. Reports, 17(1): 161–167.
64. Segal A.W. (2005) How neutrophils kill microbes. Ann. Rev. Immunol., 23: 197–223. doi: 10.1146/annurev.immunol.23.021704.115653.
65. Morimoto-Kamata R., Mizoguchi S.-I., Ichisugi T., Yui S. (2012) Cathepsin G induces cell aggregation of human breast cancer MCF-7 cells via a 2-step mechanism: Catalytic site-independent binding to the cell surface and enzymatic activity-dependent induction of the cell aggregation. Mediators Inflamm., 2012: 13. doi: 10.1155/2012/456462.456462.
66. Wilson T.J., Nannuru K.C., Futakuchi M., Singh R.K. (2010) Cathepsin G-mediated enhanced TGF-β signaling promotes angiogenesis via upregulation of VEGF and MCP-1. Cancer Letters, 288(2): 162–169. doi: 10.1016/j.canlet.2009.06.035.
67. Yui S., Osawa Y., Ichisugi T., Morimoto-Kamata R. (2014) Neutrophil cathepsin G, but not elastase, induces aggregation of MCF-7 mammary carcinoma cells by a protease activity-dependent cell-oriented mechanism.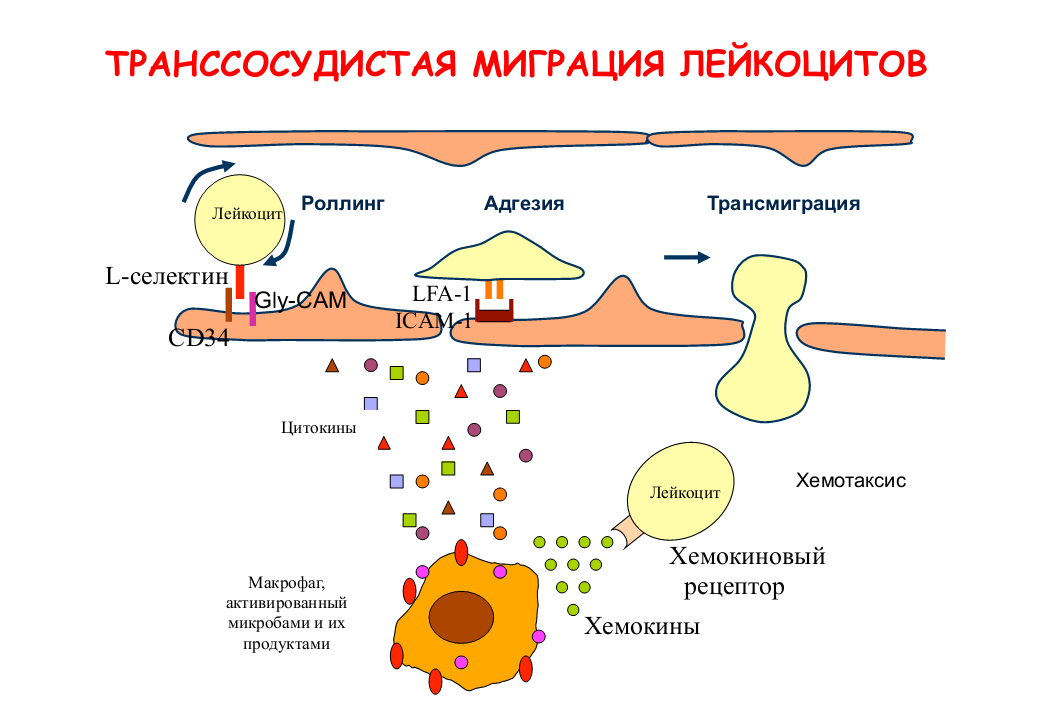 Mediators Inflamm., 2014: 12. doi: 10.1155/2014/971409.971409.
Mediators Inflamm., 2014: 12. doi: 10.1155/2014/971409.971409.
68. Acuff H.B., Carter K.J., Fingleton B. et al. (2006) Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res., 66(1): 259–266. doi: 10.1158/0008-5472.can-05-2502.
69. Ebrahem Q., Chaurasia S.S., Vasanji A. et al. (2010) Cross-talk between vascular endothelial growth factor and matrix metalloproteinases in the induction of neovascularization in vivo. Am. J. Pathol., 176(1): 496–503. doi: 10.2353/ajpath.2010.080642.
70. Hawinkels L.J.A.C., Zuidwijk K., Verspaget H.W. et al. (2008) VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer, 44(13): 1904–1913. doi: 10.1016/j.ejca.2008.06.031.
71. Coillie E.V., Aelst I.V., Wuyts A. et al. (2001) Tumor angiogenesis induced by granulocyte chemotactic protein-2 as a countercurrent principle. Am. J. Pathol., 159(4): 1405–1414. doi: 10.1016/s0002-9440(10)62527-8.
Pathol., 159(4): 1405–1414. doi: 10.1016/s0002-9440(10)62527-8.
72. Bergers G., Brekken R., McMahon G. et al. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol., 2(10): 737–744. doi: 10.1038/35036374.
73. Deryugina E.I., Zajac E., Juncker-Jensen A. et al. (2014) Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia, 16(10): 771–788. doi: 10.1016/j.neo.2014.08.013.
74. Deryugina E.I., Quigley J.P. (2015) Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biology, 44.
75. Бережная Н.М., Чехун В.Ф. (2016) Физиологическая система соединительной ткани и онкогенез. Экстрацеллюлярный матрикс и метастазирование. Онкология, 18(3): 164–176.
76. Чехун В.Ф., Бережная Н.М. (2017) Физиологическая система соединительной ткани и онкогенез. Формирование резистентности к химиопрепаратам. Онкология, 19(3): 156–170.
Онкология, 19(3): 156–170.
77. De Larco J.E., Wuertz B.R.K., Furcht L.T. (2004) The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin. Cancer Res., 10(15): 4895–4900. doi: 10.1158/1078-0432.ccr-03-0760.
78. Shabani F., McNeil J., Tippett L. (1998) The oxidative inactivation of tissue inhibitor of metalloproteinase-1 (TIMP-1) by hypochlorous acid (HOCl) is suppressed by anti-rheumatic drugs. Free Radical Res., 28(2): 115–123. doi: 10.3109/10715769809065797.
79. Cross A., Edwards S.W., Bucknall R.C., Moots R.J. (2004) Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis Rheum., 50(5): 1430–1436. doi: 10.1002/art.20166.
80. Goren I., Kämpfer H., Müller E. et al. (2006) Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: implications for normal and diabetes-impaired wounds. J. Invest. Dermatol., 126(3): 628–637. doi: 10.1038/sj. jid.5700136.
jid.5700136.
81. Grenier A., Combaux D., Chastre J. et al. (2001) Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab. Invest., 81(2): 133–141. doi: 10.1038/labinvest.3780220.
82. Grenier A., Chollet-Martin S., Crestani B. et al. (2002) Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood, 99(8): 2997–3004. doi: 10.1182/blood.v99.8.2997.
83. Matsushima A., Ogura H., Koh T. et al. (2004) Hepatocyte growth factor in polymorphonuclear leukocytes is increased in patients with systemic inflammatory response syndrome. J. Trauma, 56(2): 259–264. doi: 10.1097/01.ta.0000111752.60500.da.
84. Queen M.M., Ryan R.E., Holzer R.G. et al. (2005) Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res., 65(19): 8896–8904. doi: 10.1158/0008-5472.can-05-1734.
85. Imai Y., Kubota Y., Yamamoto S. et al. (2005) Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J. Gastroenterol. Hepatol., 20(2): 287–293. doi: 10.1111/j.1440-1746.2004.03575.x.
J. Gastroenterol. Hepatol., 20(2): 287–293. doi: 10.1111/j.1440-1746.2004.03575.x.
86. Loukinova E., Dong G., Enamorado-Ayalya I. et al. (2000) Growth regulated oncogene-alpha expression by murine squamous cell carcinoma promotes tumor growth, metastasis, leukocyte infiltration and angiogenesis by a host CXC receptor-2 dependent mechanism. Oncogene, 19(31): 3477–3486. doi: 10.1038/sj.onc.1203687.
87. Schaider H., Oka M., Bogenrieder T. et al. (2003) Differential response of primary and metastatic melanomas to neutrophils attracted by IL-8. Int. J. Cancer, 103(3): 335–343. doi: 10.1002/ijc.10775.
88. Welch D.R., Schissel D.J., Howrey R.P., Aeed P.A. (1989) Tumor-elicited polymorphonuclear cells, in contrast to ‘normal’ circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proceedings of the National Academy of Sciences of the United States of America, 86(15): 5859–5863. doi: 10.1073/pnas.86.15.5859.
89. Strell C., Lang K., Niggemann B. et al. (2010) Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Exp. Cell Res., 316(1): 138–148. doi: 10.1016/j.yexcr.2009.09.003.
Strell C., Lang K., Niggemann B. et al. (2010) Neutrophil granulocytes promote the migratory activity of MDA-MB-468 human breast carcinoma cells via ICAM-1. Exp. Cell Res., 316(1): 138–148. doi: 10.1016/j.yexcr.2009.09.003.
90. Crissman J.D., Hatfield J., Schaldenbrand M. et al. (1985) Arrest and extravasation of B16 amelanotic melanoma in murine lungs. A light and electron microscopic study. Lab. Invest., 53(4): 470–478.
91. Huh S.J., Liang S., Sharma A. et al. (2010) Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res., 70(14): 6071–6082. doi: 10.1158/0008-5472.can-09-4442.
92. Spicer J.D., McDonald B., Cools-Lartigue J.J. et al. (2012) Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res., 72(16): 3919–3927. doi: 10.1158/0008-5472.CAN-11-2393.
93. Kousis P.C., Henderson B.W., Maier P.G., Gollnick S.O. (2007) Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res., 67(21): 10501–10510. doi: 10.1158/0008-5472.can-07-1778.
Cancer Res., 67(21): 10501–10510. doi: 10.1158/0008-5472.can-07-1778.
94. Suttmann H., Riemensberger J., Bentien G. et al. (2006) Neutrophil granulocytes are required for effective Bacillus Calmette-Guérin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res., 66(16): 8250–8257. doi: 10.1158/0008-5472.can-06-1416.
95. Huang S., Mills L., Mian B. et al. (2002) Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am. J. Pathol., 161(1): 125–134. doi: 10.1016/s0002-9440(10)64164-8.
96. Otten M.A., Leusen J.H.W., Rudolph E. et al. (2007) FcR γ-chain dependent signaling in immature neutrophils is mediated by FcαRI, but not by FcγRI. J. Immunol., 179(5): 2918–2924. doi: 10.4049/jimmunol.179.5.2918.
97. Bakema J.E., Ganzevles S.H., Fluitsma D.M. et al. (2011) Targeting FcαRI on polymorphonuclear cells induces tumor cell killing through autophagy. J. Immunol..gif) , 187(2): 726–732. doi: 10.4049/jimmunol.1002581.
, 187(2): 726–732. doi: 10.4049/jimmunol.1002581.
98. Uribe-Querol E., Rosales C. (2015) Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res., 2015: 983698. Pub. online 2015 Dec. 24. doi: 10.1155/2015/983698.
Адрес:
Лисяный Николай Иванович
04050, Киев, ул. П. Майбороды, 32
ГУ «Институт нейрохирургии
им. акад. А.П. Ромоданова НАМН Украины»
Тел.: (044) 483-01-93
E-mail: [email protected]
Correspondence:
Lisyaniy Mykola
32 P. Mayborody str., Kyiv 04050
SI «Institute of Neurosurgery named after acad. A.P. Romodanov
NAMS of Ukraine»
Tel.: 044 483-01-93
E-mail: [email protected]
(PDF) НЕЙТРОФИЛЬНЫЕ ВНЕКЛЕТОЧНЫЕ ЛОВУШКИ (Neutrophil Extracellular Traps NETS)
2
Спустя столетие после описания фагоцитоза Ильёй Мечниковым, Zychlinsky Α. и его
коллеги, в 2004 году, описали новый антимикробный механизм нейтрофильных
гранулоцитов: образование и высвобождение нейтрофильных внеклеточных ловушек
(НВЛ) или сетей (neutrophil extracellular traps (NETs) [2].
NETs представляют собой фибриллярную структуру, «скелет» которой состоит из
деконцентрированного хроматина, то есть ядерной ДНК и гистонов. В процессе
образования NETs, происходит связывание хроматина с различными ферментами и белками
гранул нейтрофилов, например нейтрофильной эластазой (NE) и миелопероксидазой (MPO)
[3]. Следует отметить, что белковый компонент внеклеточных нейтрофильных ловушек
непостоянен и определяется микросредой, непосредственно окружающей клетки [4].
Например, у людей с острым инфарктом миокарда нейтрофилы синтезируют тканевой
фактор (TF), один из ключевых факторов тромбообразования, который при образовании
внеклеточных нейтрофильных ловушек в очаге разрыва уязвимой атеросклеротической
бляшки активирует тромбоциты и способствует окклюзии сосудов [5].
Нейтрофилы, после высвобождения NETs, подвергаются клеточной гибели, которая
отличается от апоптоза и некроза и называется НЕТоз. В отличие от некроза и апоптоза,
В отличие от некроза и апоптоза,
при нетозе не наблюдается фрагментации ДНК и отсутствуют специфические молекулы,
необходимые для «очистки» продуктов гибели клеток макрофагами.
Механизмы формирования / регулирования NETs
Молекулярные механизмы, участвующие в образовании, высвобождении и клиренсе
NETs, полностью не выяснены. Наиболее изученные механизмы это индукция аутофагии и
гиперцитруллинизация гистонов ферментом пептидиларгининдеминаза-4 (PAD4).
Установлено, что активные формы кислорода (АФК) также участвуют в образовании NETs.
Гомеостатически, клиренс NETs достигается за счет внеклеточной деградации ферментом
ДНКазы и / или их фагоцитоз макрофагами [1,4].
Hейтрофильные внеклеточные ловушки и инфекции
Индукция формирования NET сетей является основным механизмом естественного
иммунитета. Процесс запускается широким спектром патогенных микроорганизмов, как
грамположительных бактерий (Staphylococcus aureus, Staphylococcus suis, Streptococcus
pyogenes, Streptococcus pneumoniae), так и грамотрицательных бактерий (Escherichia coli,
Salmonella enterica, Shigella flexneri, Pseudomonas aerugin), дрожжами (Candida albicans) и
простейшими паразитами (Leishmania amazonensis или Trypanosoma cruzi). Во
Во
внеклеточных нейтрофильных ловушках микроорганизмы фиксируются, предотвращается
распространение инфекции и подвергаются воздействию ферментов и антибактериальных
белков, таких как LL 37, которые воздействуют на микроорганизмы локально и в высоких
концентрациях [6].
Важно отметить, что микроорганизмы в своей попытке выжить, разработали
механизмы ингибирования NETs. Такими механизмами являются:
Синтез полисахаридной капсулы (Streptococcus pneumoniae)
Формирование биомембран (Pseudomonas aeruginosa)
Изменение поверхностного заряда микроорганизмов (Streptococcus группы А, Shigella
flexneri)
Следует отметить, что наряду с микробной стимуляцией, образование ловушек также
может быть обусловлен различными сигналами, включая фармакологические средства,
воспалительные цитокины, фактор некроза опухоли альфа, ФНО-α, иммунные комплексы,
силиконовые имплантанты и другие.
Hейтрофильные внеклеточные ловушки и сепсис
Нейтрофил — это… Что такое Нейтрофил?
Нейтрофильные гранулоциты или нейтрофилы, сегментоядерные нейтрофилы, нейтрофильные лейкоциты — подвид гранулоцитарных лейкоцитов, названный нейтрофилами за то, что при окраске по Романовскому они интенсивно окрашиваются как кислым красителем эозином, так и основными красителями, в отличие от эозинофилов, окрашиваемых только эозином, и от базофилов, окрашиваемых только основными красителями.
Зрелые нейтрофилы имеют сегментированное ядро, то есть относятся к полиморфноядерным лейкоцитам, или полиморфонуклеарам.
Зрелые сегментоядерные нейтрофилы в норме являются основным видом лейкоцитов, циркулирующих в крови человека, составляя от 47% до 72% общего количества лейкоцитов крови. Еще 1-5% в норме составляют юные, функционально незрелые нейтрофилы, имеющие палочкообразное сплошное ядро и не имеющие характерной для зрелых нейтрофилов сегментации ядра — так называемые палочкоядерные нейтрофилы.
Нейтрофилы способны к активному амебоидному движению, к экстравазации (эмиграции за пределы кровеносных сосудов), и к хемотаксису (преимущественному движению в направлении мест воспаления или повреждения тканей).
Нейтрофилы способны к фагоцитозу, причём являются микрофагами, то есть способны поглощать лишь относительно небольшие чужеродные частицы или клетки. После фагоцитирования чужеродных частиц нейтрофилы обычно погибают, высвобождая большое количество биологически активных веществ, повреждающих бактерии и грибки, усиливающих воспаление и хемотаксис иммунных клеток в очаг. Нейтрофилы содержат большое количество миелопероксидазы, фермента, который способен окислять анион хлора до гипохлорита — сильного антибактериального агента. Миелопероксидаза как гем-содержащий белок имеет зеленоватый цвет, что определяет зеленоватый оттенок самих нейтрофилов, цвет гноя и некоторых других выделений, богатых нейтрофилами. Погибшие нейтрофилы вместе с клеточным детритом из разрушенных воспалением тканей и гноеродными микроорганизмами, послужившими причиной воспаления, формируют массу, известную как гной.
Повышение процента нейтрофилов в крови называется относительным нейтрофилезом, или относительным нейтрофильным лейкоцитозом. Повышение абсолютного числа нейтрофилов в крови называется абсолютным нейтрофилезом. Снижение процента нейтрофилов в крови называется относительной нейтропенией. Снижение абсолютного числа нейтрофилов в крови обозначается как абсолютная нейтропения.
Нейтрофилы играют очень важную роль в защите организма от бактериальных и грибковых инфекций, и сравнительно меньшую — в защите от вирусных инфекций. В противоопухолевой или антигельминтной защите нейтрофилы практически не играют роли.
Нейтрофильный ответ (инфильтрация очага воспаления нейтрофилами, повышение числа нейтрофилов в крови, сдвиг лейкоцитарной формулы влево с увеличением процента «юных» форм, указывающий на усиление продукции нейтрофилов костным мозгом) — самый первый ответ на бактериальные и многие другие инфекции. Нейтрофильный ответ при острых воспалениях и инфекциях всегда предшествует более специфическому лимфоцитарному. При хронических воспалениях и инфекциях роль нейтрофилов незначительна и преобладает лимфоцитарный ответ (инфильтрация очага воспаления лимфоцитами, абсолютный или относительный лимфоцитоз в крови).
При хронических воспалениях и инфекциях роль нейтрофилов незначительна и преобладает лимфоцитарный ответ (инфильтрация очага воспаления лимфоцитами, абсолютный или относительный лимфоцитоз в крови).
Wikimedia Foundation. 2010.
Роль нейтрофилов в развитии полиорганной недостаточности при сепсисе Текст научной статьи по специальности «Фундаментальная медицина»
РОЛЬ НЕИТРОФИЛОВ В РАЗВИТИИ ПОЛИОРГАННОЙ НЕДОСТАТОЧНОСТИ ПРИ СЕПСИСЕ
ROLE OF NEUTROPHILS IN DEVELOPMENT OF MULTIPLE ORGAN FAILURE IN SEPSIS
Устьянцева И.М. Хохлова О.И.
Ustyantseva I.M. Khokhlova O.I.
Федеральное государственное Federal State Medical Prophylactic Institution
лечебно-профилактическое учреждение «Scientific Clinical Center of Miners’
«Научно-клинический центр охраны здоровья шахтеров», Health Protection»,
г. Ленинск-Кузнецкий, Россия Leninsk-Kuznetsky, Russia
Ленинск-Кузнецкий, Россия Leninsk-Kuznetsky, Russia
Полиорганная недостаточность является серьезной угрозой для выживания пациентов с сепсисом и системным воспалением. Иммунная система противостоит микробным инфекциям, но при тяжелом сепсисе ее неблагоприятная активность приводит к развитию органной дисфункции. Патологическим проявлениям полиорганной недостаточности способствует несоответствующая активация и размещение нейтрофилов в пределах микроциркуляторного русла.
Ключевые слова: полиорганная недостаточность, сепсис, нейтрофилы.
Multiple organ failure is a major threat to the survival of patients with sepsis and systemic inflammation. The immune system combats microbial infections but, in severe sepsis, its untowards activity seems to contribute to organ dysfunction. An inappropriate activation and positioning of neutrophils within the microvasculature contributes to the pathological manifestations of multiple organ failure.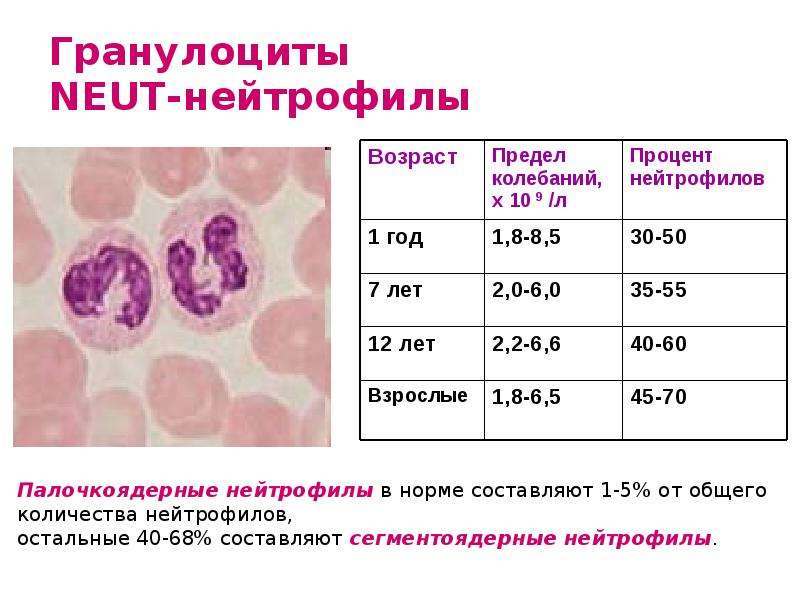
Key words: multiple organ failure, sepsis, neutrophils.
Согласно статистическим данным США и Европейского Союза, сепсис является одной из ключевых клинических проблем, что обусловлено высокой смертностью (до 28-50 %) [2]. Ежедневно в мире от сепсиса умирают более 1400 человек [1, 2].
Сепсис определен как системная воспалительная реакция на инфекцию с его тяжелой формой, связанной с признаками органной дисфункции. В патогенез органной дисфункции вовлечены несколько факторов: эндокринная и иммунная системы, диссеминированное внут-рисосудистое свертывание, генетическая предрасположенность, нарушение энергетического метаболизма.
Первоначальными факторами системного воспаления при сепсисе часто являются бактериальные компоненты, которые стимулируют се-
Корреспонденцию адресовать: Устьянцева Ирина Марковна, д. б.н., профессор 7-й микрорайон, № 9, ФГЛПУ «НКЦОЗШ», г. Ленинск-Кузнецкий, Кемеровская область, 652509, Россия Тел.: 8 (38456) 2-38-88, 9-55-13 E-mail: [email protected]
б.н., профессор 7-й микрорайон, № 9, ФГЛПУ «НКЦОЗШ», г. Ленинск-Кузнецкий, Кемеровская область, 652509, Россия Тел.: 8 (38456) 2-38-88, 9-55-13 E-mail: [email protected]
крецию провоспалительных цито-кинов, таких как интерлейкин 1р, интерлейкин-6 и фактор некроза опухолей а (ФНО а). Первичная цитокинемия сопровождается компенсаторной реакцией повышенных концентраций циркулирующих антивоспалительных цитокинов (ин-терлейкин-10).
Нейтрофилы играют основную роль в защите против микробных инфекций, что связано с наличием большого количества протеолити-ческих ферментов и быстрой продукцией активных форм кислорода для уменьшения внутренних патогенов. Если эти литические факторы или провоспалительные цитоки-ны высвобождаются внеклеточно из инфильтрующих ткань нейтро-филов, результатом будет являться локальное повреждение. При тяжелом сепсисе локальная инфекция сопровождается системной активацией нейтрофилов.![]()
Исследование аутопсических образцов от пациентов с полиорганной недостаточностью показывает локализацию нейтрофилов, варьирующую от секвестрации и агрегации в почечных кровеносных сосудах до широкой тканевой инфильтрации легкого [3]. При остром респираторном дистресс-синдроме, более тяжелой форме острого по-
вреждения легких, которое может сопровождать сепсис, плотность нейтрофильных инфильтратов соотносится с нарушенной функцией легких и с высокими концентрациями в бронхоальвеолярной жидкости протеолитических ферментов, источником которых являются ней-трофилы [17].
При системном воспалении гоме-остатические механизмы в микро-циркуляторном русле осложняются эндотелиальной гиперактивностью, фибриновым депонированием, закупоркой сосудов и, иногда, появлением тканевых экссудатов, что в дальнейшем препятствует соответствующей оксигенации. Нейтрофи-лы участвуют в этих реологических изменениях через их усиленное связывание со стенками кровеносных сосудов и через формирование лейкоцитарных скоплений. Имеются факты, показывающие, что активность нейтрофилов может быть причинно-обусловленной, о чем свидетельствует повышение респираторной и почечной функции у пациентов с системным воспалением при удалении нейтрофилов из кровеносного русла с помощью лейкоснижающих фильтров [13]. При этом нейтрофилы, удержанные фильтрами, предрасположены к эндотелиальному связыванию,
Нейтрофи-лы участвуют в этих реологических изменениях через их усиленное связывание со стенками кровеносных сосудов и через формирование лейкоцитарных скоплений. Имеются факты, показывающие, что активность нейтрофилов может быть причинно-обусловленной, о чем свидетельствует повышение респираторной и почечной функции у пациентов с системным воспалением при удалении нейтрофилов из кровеносного русла с помощью лейкоснижающих фильтров [13]. При этом нейтрофилы, удержанные фильтрами, предрасположены к эндотелиальному связыванию,
№ 2 [июнь] 2008
71
что указывает на связь между взаимодействием нейтрофилов со стенками кровеносных сосудов и органной дисфункцией. Данные по животным моделям совпадают с клиническими наблюдениями. Большое количество нейтрофилов накапливается в органах, испытывающих недостаточность, а травма органа может стимулировать приток и секвестрацию нейтрофилов в других органах с последующей полиорганной дисфункцией [10].
ФИЗИОЛОГИЯ И ПАТОФИЗИОЛОГИЯ
Нейтрофилы развиваются в костном мозге за 14 дней и временно остаются в пуле хранения перед выбросом в кровь, где они в течение 12-14 часов переходят из пула циркулирующих клеток (осевой поток) в пул скопления лейкоцитов (контакт со стенками кровеносных сосудов). Далее, при отсутствии бактериальных инфекций, нейтро-филы поступают в ретикулоэндо-телиальные органы, например, в печень, или даже возвращаются в костный мозг, чтобы подвергнуться апоптозу. Стареющие нейтрофилы превращаются в апоптические тела, которые достигают высшей ступени при фагоцитозе локальными макрофагами, предотвращая таким образом появление повреждения тканей литическими факторами, высвобождаемыми из стареющих клеток.
Устранение нейтрофилов через апоптоз является гомеостатиче-ским механизмом, который предотвращает повреждение здоровых тканей.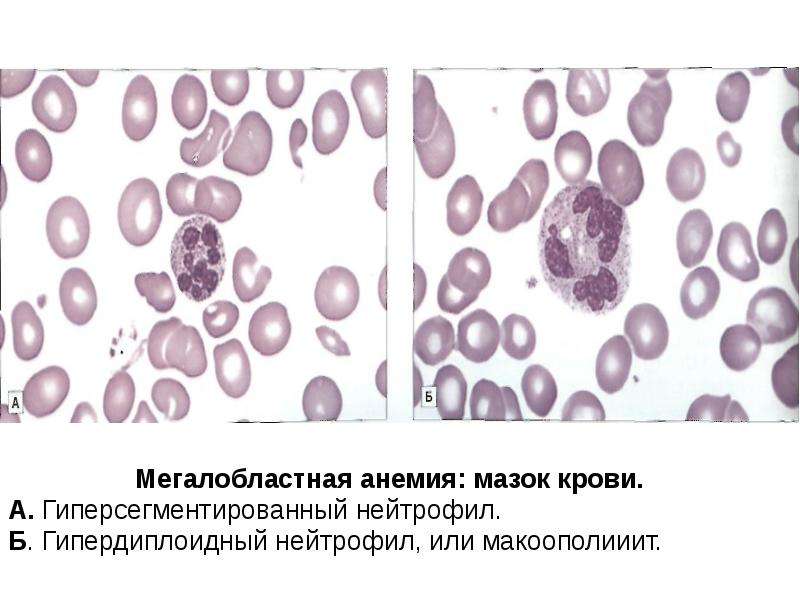 Этот процесс является центральным в профилактике и разрешении воспаления. У пациентов с системным воспалением, системными инфекциями, тяжелым сепсисом и риском полиорганной недостаточности [15] апоптоз ней-трофилов подавлен, что происходит вследствие активности циркулирующих факторов (липополи-сахарид, липотейхоевая кислота и провоспалительные цитокины). При этом связывание нейтрофи-лов с эндотелием, активированным провоспалительными цитокинами, увеличивает продолжительность жизни нейтрофилов по сравнению с нестимулированным эндотелием, который ускоряет смерть клеток.
Этот процесс является центральным в профилактике и разрешении воспаления. У пациентов с системным воспалением, системными инфекциями, тяжелым сепсисом и риском полиорганной недостаточности [15] апоптоз ней-трофилов подавлен, что происходит вследствие активности циркулирующих факторов (липополи-сахарид, липотейхоевая кислота и провоспалительные цитокины). При этом связывание нейтрофи-лов с эндотелием, активированным провоспалительными цитокинами, увеличивает продолжительность жизни нейтрофилов по сравнению с нестимулированным эндотелием, который ускоряет смерть клеток.
При сепсисе выживание нейтро-филов в ткани может быть в дальнейшем увеличено за счет действия местных антиапоптотических факторов. Это наблюдается при остром респираторном дисстресс-синдро-ме, при котором низкий уровень апоптоза альвеолярных нейтрофи-лов связан с концентрацией интер-лейкина-2 в бронхоальвеолярной жидкости [Lesuг]. Ингибирование апоптоза происходит через дисре-гуляцию сложной сети внутриклеточной сигнализации и функций органелл. Большая продолжительность жизни нейтрофилов у пациентов с сепсисом контрастирует с повышенным апоптозом лимфоцитов в лимфоидной ткани и последующим иммунопараличом [7].
Большая продолжительность жизни нейтрофилов у пациентов с сепсисом контрастирует с повышенным апоптозом лимфоцитов в лимфоидной ткани и последующим иммунопараличом [7].
Критерии диагностики сепсиса, как известно, включают в себя количество нейтрофилов: высокое (более 12 х 109/л), низкое (менее 4 х 109/л) или нормальное, но при наличии более 10 % незрелых клеток. Высокие концентрации кровяных нейтрофилов могут быть связаны с излишней продукцией костным мозгом, возвращением клеток маргинации в пул циркулирующих клеток, подавлением апоптоза или и с тем, и с другим.
Гранулоцитарный колониести-мулирующий фактор (ГКФ) и колониестимулирующий грануло-цитарно-макрофагальный фактор (КГМФ) увеличивают количество циркулирующих нейтрофилов, стимулируют их созревание и активацию и увеличивают продолжительность жизни нейтрофилов. У здоровых людей концентрация ГКФ в крови очень низкая, тогда как в острую стадию инфекции наблюдается ее увеличение в несколько раз с последующим повышением числа нейтрофилов.
Имеются данные, показывающие, что большую патогенную роль при тяжелом сепсисе несет не общее количество нейтрофилов в кровотоке, а наличие клеточной подгруппы, чей фенотип и уровень активации стимулируют повреждение тканей [11]. В животных моделях сепсиса незрелые нейтрофилы преимущественно накапливаются в микрососудах легких, где их активация стимулирует существенное повреждение тканей через
высвобождение протеолитических ферментов [9].
Предполагается, что воздействие нейтрофилов на патологию может возникнуть из-за наличия популяций нейтрофилов, которые предрасположены к эндотелиальному взаимодействию. Иными словами, нейтрофилы не являются функционально гомогенными, они состоят из субпопуляций с определенными фенотипическими и секреторными параметрами.
Нейтрофилы существуют в трех состояниях: покоя (нестимулиро-ванное), возбужденном (столкновение с воспалительным агонистом или веществом, ослабляющим пороговые раздражители, необходимые для активации) и активированном (прохождение определенной функции). Переход нейтрофилов из состояния покоя в кровообращении к активированному состоянию в участке инфекции выполняется заданной последовательностью сигналов от мотивирующих раздражителей (комплементный пептид С5а, липополисахарид, цитокины).
Переход нейтрофилов из состояния покоя в кровообращении к активированному состоянию в участке инфекции выполняется заданной последовательностью сигналов от мотивирующих раздражителей (комплементный пептид С5а, липополисахарид, цитокины).
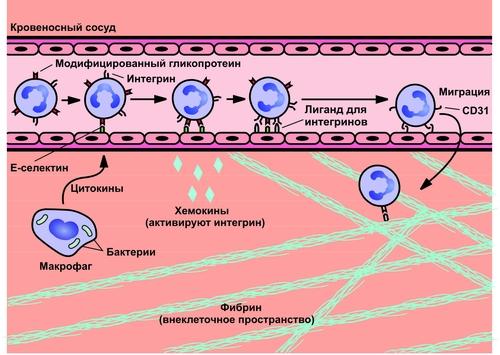
Бактериальная ликвидация зависит от быстрого поступления кровяных нейтрофилов в участки инфекции. При этом нейтрофилы, в первую очередь, должны присоединиться к стенкам кровеносного сосуда, прежде чем активно мигрировать в окружающую ткань в ответ на химические раздражители (хемотаксис). Связывание нейтро-филов с сосудистым эндотелием контролируется последовательной активностью двух групп молекул адгезии, селектинами и интегрина-ми. Селектины вызывают первичное прикрепление нейтрофилов к эндотелию при касательном усилии кровотока, тогда как интегрины вызывают плотную адгезию.
ХЕМОТАКСИС
Кровяные нейтрофилы отвечают на хемотаксический фактор, высвобождаемый в источнике инфекции, например, комплементный пептид С5а, лейкотриен В, фактор активации тромбоцитов, бактериальный пептид формил-метионил-лейко-фенилаланин и интерлейкин-8.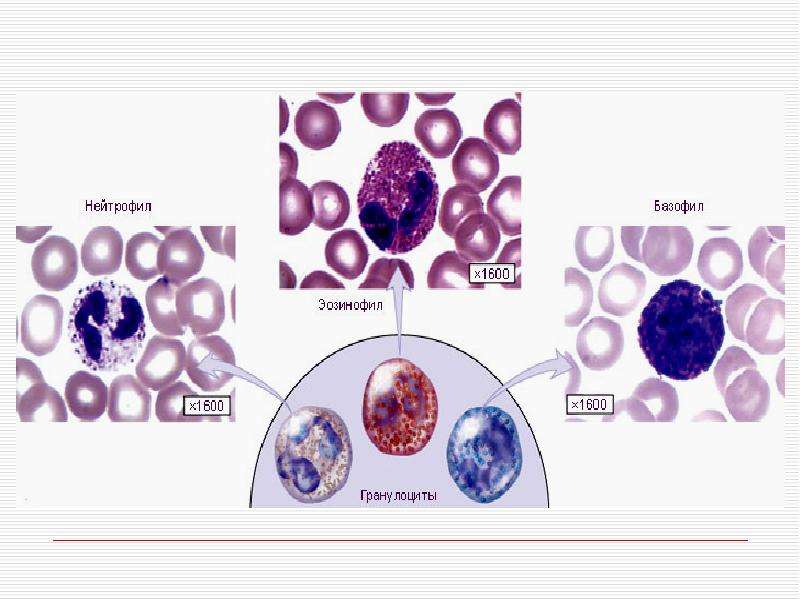 Клетки мигрируют из области низкой концентрации (т.е. стенки кровеносных сосудов) в область
Клетки мигрируют из области низкой концентрации (т.е. стенки кровеносных сосудов) в область
ПОЛИТРАВМА
высокой концентрации (участок инфекции или воспаления), вследствие чего хемотаксические факторы становятся мощными активаторами нейтрофилов.
У пациентов с острым респираторным дистресс-синдромом нейтрофилы проходят крупномасштабную миграцию в легкие. В большом круге кровообращения нейтрофилы входят в ткань через посткапиллярные венулы, но в малом круге миграция происходит через капилляры [16]. Нейтрофи-лы могут быстро войти в легкое, в отличие от других органов, из-за отличительного фенотипа капиллярного эндотелия и из-за ответной реакции подгруппы нейтрофилов на хемотаксические факторы или микроорганизмы, связанные с легочной инфекцией [12].
Циркулирующие факторы (ин-терлейкин-8, ФНОа и др.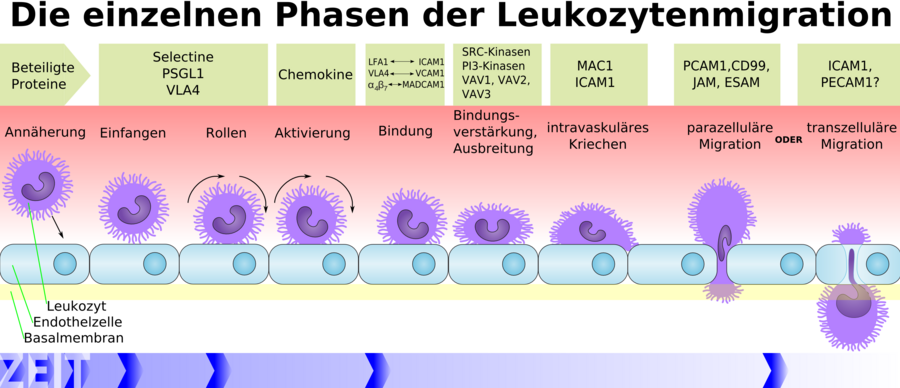 ) могут также отвечать за функциональный статус кровяных нейтрофилов при сепсисе. Увеличивая продолжительность существования нейтро-филов и подавляя их миграцию по сосудистой сети, циркулирующие факторы повышают нейтрофил-эн-дотелиальные клеточные взаимодействия и усиливают повреждение сосудов. То есть, дисфункция ней-трофилов при тяжелом сепсисе не является первичным механизмом, но является последствием системной активации.
) могут также отвечать за функциональный статус кровяных нейтрофилов при сепсисе. Увеличивая продолжительность существования нейтро-филов и подавляя их миграцию по сосудистой сети, циркулирующие факторы повышают нейтрофил-эн-дотелиальные клеточные взаимодействия и усиливают повреждение сосудов. То есть, дисфункция ней-трофилов при тяжелом сепсисе не является первичным механизмом, но является последствием системной активации.
РАСПОЗНАВАНИЕ
И ФАГОЦИТОЗ БАКТЕРИЙ
Нейтрофилы у пациентов с сепсисом обладают повышенной ин-тернализацией и способностью к деструкции микроорганизмов [15].
Нейтрофильное связывание бактерий значительно повышается, когда патогены покрываются IgG. Высокоаффинный рецептор для IgG — это CD64, который обычно не определяется в интактных ней-трофилах и считается маркером активированных нейтрофилов.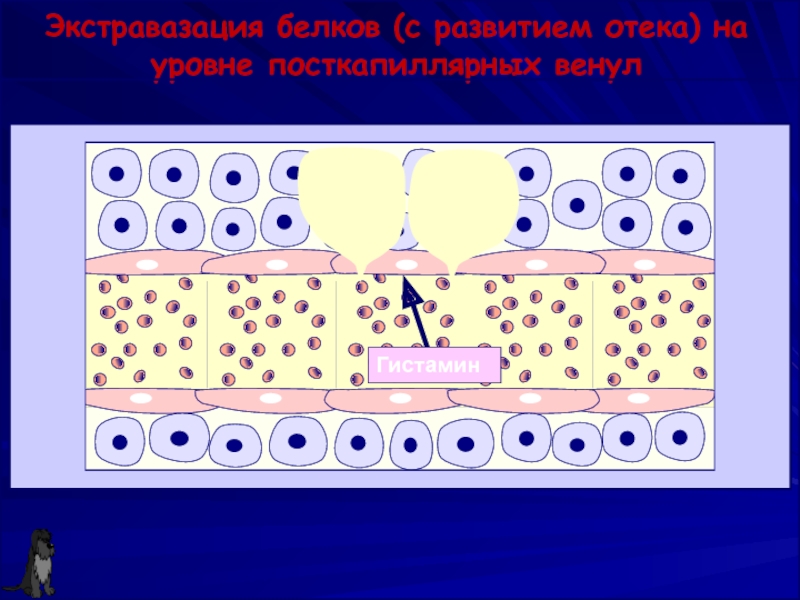 Его экспрессия стимулируется интерфероном у и КГМФ. Большинство нейтрофилов у пациентов с сепсисом экспрессирует CD64, а сверхрегуляция CD64 на неонатальных нейтрофилах считается показателем сепсиса. Связывание с бактериями происходит также через CD14, ре-
Его экспрессия стимулируется интерфероном у и КГМФ. Большинство нейтрофилов у пациентов с сепсисом экспрессирует CD64, а сверхрегуляция CD64 на неонатальных нейтрофилах считается показателем сепсиса. Связывание с бактериями происходит также через CD14, ре-
цептор липополисахарида, который в норме присутствует на всех моноцитах. Этот рецептор слабо выражен на нейтрофилах, но становится сверхрегулируемым в ответ на бактериальные инфекции [5]. Другие рецепторы, которые экспрессиру-ются на нейтрофилах у пациентов с сепсисом, усиливающие фагоцитоз и бактериальное распознавание, включают рецептор С3Ь (связывает комплементный пептид СЗЬ), CD16 и CD32 (подобны CD64, также связывают Fc участки IgG).
То11-подобные рецепторы являются рецепторами распознавания структур, которые контролируют наследственные иммунные реакции на различные микробные лиганды. На сегодняшний день существует 11 форм. То11-подобный рецептор 4, который тесно связан с CD14, является сигнальным трансдуци-рующим рецептором липополиса-харида, тогда как То11-подобный рецептор 2 преимущественно распознает граммпозитивные бактерии. Нестимулированные нейтрофилы у здоровых людей содержат некоторые из двух типов рецепторов на своей поверхности, хотя То11-по-добный рецептор 2 сверхрегулиру-ется гранулоцитарным колониести-мулирующим фактором (ГКФ) и колониестимулирующим грануло-цитарно-макрофагальным фактором (КГМФ). Фармакологическая модификация То11-подобных рецепторов выдвигается как будущая терапевтическая стратегия для пациентов септическим шоком [4].
То11-подобный рецептор 4, который тесно связан с CD14, является сигнальным трансдуци-рующим рецептором липополиса-харида, тогда как То11-подобный рецептор 2 преимущественно распознает граммпозитивные бактерии. Нестимулированные нейтрофилы у здоровых людей содержат некоторые из двух типов рецепторов на своей поверхности, хотя То11-по-добный рецептор 2 сверхрегулиру-ется гранулоцитарным колониести-мулирующим фактором (ГКФ) и колониестимулирующим грануло-цитарно-макрофагальным фактором (КГМФ). Фармакологическая модификация То11-подобных рецепторов выдвигается как будущая терапевтическая стратегия для пациентов септическим шоком [4].
Еще одна группа рецепторов распознавания — инициирующий рецептор, экспрессируемый на ми-елоидных клетках (ИРВМК). При инфицировании бактериями отмечается инфильтрация ткани нейтро-филами с высокими концентрациями ИРВМК-1. Сигнализация через ИРВМК-1 высвобождает интерлей-кин-8, сверхрегулирует поверхностные адгезионные молекулы. Представление, что ИРВМК-1 может играть существенную патогенную роль при сепсисе, поддерживается сведениями, показывающими, что повышенная экспрессия на нейтро-филах ограничена острыми воспалительными реакциями, которые ускоряют инфекционные агенты, и что переход в растворимую форму
Представление, что ИРВМК-1 может играть существенную патогенную роль при сепсисе, поддерживается сведениями, показывающими, что повышенная экспрессия на нейтро-филах ограничена острыми воспалительными реакциями, которые ускоряют инфекционные агенты, и что переход в растворимую форму
ИРВМК-1 у пациентов с сепсисом приводит к благоприятным результатам [14].
НЕЙТРОФИЛ-
ЭНДОТЕЛИАЛЬНЫЕ
ВЗАИМОДЕЙСТВИЯ
И СВЯЗАННАЯ С
СЕПСИСОМ ОРГАННАЯ
НЕДОСТАТОЧНОСТЬ
При тяжелом сепсисе присутствует функциональная дихотомия ней-трофилов, связанная с ответной реакцией на бактериальные инфекции. В нелегочной ткани экстравазация нейтрофилов в участки инфекции затруднена, возможно, из-за излишнего эндотелиального связывания и сниженной хемотаксической отвеча-емости, в противоположность интенсивной инфильтрации этих клеток в инфицированную легочную ткань. Секвестрация нейтрофилов может быть ключевой стадией в инициации полиорганной недостаточности. Связывание нейтрофилов со стенками кровеносных сосудов может быть опосредовано абнормальной экспрессией молекул адгезии, отличных от стимулирующего прикрепления к легочному эндотелию, или молекулами, имеющими высокую авидность для соответствующих эндотелиальных лигандов. Повышенная адгезия, в дальнейшем спровоцированная деактивацией хемотаксических рецепторов, может продуцировать микрососудистые окклюзии с последующей тканевой гипоперфузией и гипоксией [6]. Если нейтрофил-эндотелиальное связывание происходит в микрососудистых участках, то возможно образование лейко-агрегатов. Более того, если нейтрофилы, стимулированные циркулирующими факторами, сталкиваются с дополнительными стимулирующими агентами на стенке кровеносного сосуда, то эти нейтрофилы могут высвобождать литические факторы, что приводит к повреждению эндотелиальных клеток и повышает проницаемость сосудов.
В нелегочной ткани экстравазация нейтрофилов в участки инфекции затруднена, возможно, из-за излишнего эндотелиального связывания и сниженной хемотаксической отвеча-емости, в противоположность интенсивной инфильтрации этих клеток в инфицированную легочную ткань. Секвестрация нейтрофилов может быть ключевой стадией в инициации полиорганной недостаточности. Связывание нейтрофилов со стенками кровеносных сосудов может быть опосредовано абнормальной экспрессией молекул адгезии, отличных от стимулирующего прикрепления к легочному эндотелию, или молекулами, имеющими высокую авидность для соответствующих эндотелиальных лигандов. Повышенная адгезия, в дальнейшем спровоцированная деактивацией хемотаксических рецепторов, может продуцировать микрососудистые окклюзии с последующей тканевой гипоперфузией и гипоксией [6]. Если нейтрофил-эндотелиальное связывание происходит в микрососудистых участках, то возможно образование лейко-агрегатов. Более того, если нейтрофилы, стимулированные циркулирующими факторами, сталкиваются с дополнительными стимулирующими агентами на стенке кровеносного сосуда, то эти нейтрофилы могут высвобождать литические факторы, что приводит к повреждению эндотелиальных клеток и повышает проницаемость сосудов.
Органная недостаточность может быть результатом воздействия нейтрофилов на функцию эндо-телиальных клеток. Нейтрофилы являются важным источником про-воспалительных цитокинов. Секреция цитокинов нейтрофилами, свя-
№ 2 [июнь] 2008
занными со стенками кровеносных сосудов, может изменить нетром-богенные свойства эндотелия до прокоагулянтного состояния с активацией диссеминированной вну-трисосудистой коагуляции, а также стимулировать продукцию окиси азота в эндотелиальных и гладких мышечных клетках. Помимо развития гипотензии септического шока, высвобождение окиси азота может нарушить метаболизм в тканях через ингибирование митохондриаль-ных ферментов. В легких органная дисфункция обусловлена деструкцией базальной мембраны альвеол и повреждением типичного механизма альвеолярного жидкостного клиренса [6]. Большое количество стимулированных нейтрофилов, входящих в альвеолярную ткань и пространства, выделяет протео-литические ферменты и радикалы кислорода в ответ на неблагоприятную активацию локальными воспалительными факторами или бактериальными продуктами.
Литература:
Таким образом, доказано, что при сепсисе подавляется иммунная реакция, что отражается в гипоре-активности лимфоцитов и уменьшении их количества вследствие повышенного апоптоза. Напротив, нейтрофилы при сепсисе участвуют в отвержении инвазивных агентов, одновременно стимулируя сопутствующее повреждение, при котором поражается органная функция. Раскрыты механизмы повреждающего действия нейтрофилов. Тем не менее, разъяснение функционального статуса нейтрофилов у пациентов с сепсисом ограничено недостаточными исследованиями в некоторых областях (в частности, фагоцитоз) и противоположными данными в других (экспрессия молекул адгезии). Несоответствия, вероятно, связаны с неадекватной стратификацией пациентов, применением различных лекарств (например, стероидов) и различиями в экспериментальном проекте. Значимая оценка поведения нейтрофилов при
сепсисе нуждается в динамических исследованиях индивидуальных пациентов, т.к. у нейтрофилов короткий период полужизни в кровотоке, а по одной временной точке трудно объяснить расстройство, которое длится днями или неделями. Аспекты типичного жизненного цикла нейтрофилов все еще не полностью поняты. Например, мало что известно о механизмах производства ГКФ и КГМФ и высвобождения нейтро-филов из костного мозга, и зависит ли экстравазация в апоптотические органы от тех же поверхностных адгезионных молекул и хемотакси-ческих рецепторов, стимулирующих направленную миграцию в участки инфекции и воспаления. Тем не менее, имеющиеся данные позволяют предположить, что выборочное нацеливание на взаимодействие подгрупп нейтрофилов со стенками кровеносных сосудов в органах, подверженных дисфункции, могут представлять ценность для дальнейшего терапевтического поиска.
1. Мальцева, Л.А. Сепсис: эпидемиология, патогенез, диагностика, интенсивная терапия /Л.А. Мальцева, Л.В. Усенко, Н.Ф. Мосенцев /под ред. Л.В. Усенко. — Донецк: АРТ-ПРЕСС, 2004. — С. 10-14.
2. Angus, D.C. Epidemiology of sepsis: an update /D.C. Angus, R.S. Wax //Crit. Care Med. — 2001. — Vol. 29. — P. 109-116.
3. Brealey, D. Multi-organ dysfunction in the critically ill: effects on different organs /D. Brealey, M. Singer //J. R. Coll. Physn. Lon.
— 2000. — Vol. 34. — P. 428-436.
4. Cristorfaro, P. The TOLL-like receptors and their role in septic shock /P. Cristorfaro, S.M. Opal //Expert Opin. Ther. Targets. — 2003.
— Vol. 7. — P. 603-612.
5. Expression patterns of the lipopolysaccharide receptor CD14, and the FCy receptors CD16 and CD 64 on polymorphonuclear neutrophils: data from patients with severe Bacterial infections and lipo-polysaccharide-exposed cells /C. Wagner, R. Deppisch, B. Denefleh et al. //Shock. — 2003. — Vol. 19. — P. 5-12.
6. Fink, M.P. Mechanisms of organ Dysfunction in critical illness: report from a Round Table Conference held in Brussels /M.P. Fink, T.W. Evans //Int. Care Med. — 2002. — Vоl. 28. — P. 369-375.
7. Hotchkiss, R.S. The pathophysiology and treatment of sepsis /R.S. Ho-tchkiss, I.E. Karl //N. Engl. J. Me. — 2003. — Vol. 348. — P. 138-150.
8. Interleukin-2 involvement in early acute respiratory distress syndrome: relationship with polymorphonucler neutrophil apoptosis and patient suevival /O. Lesur, A. Kokis, C. Hermans et al. //Crit. Care Med. — 2000. — Vol. 29. — P. 3814-3822.
9. Neutrophil defensis mediate acute inflfmmatory response and lung dysfunction in dose-related fashion /H. Zhang, G. Porro, N. Orzech //Am. J. Physiol. Lung Cell Mol. Physiol. — 2001. — Vol. 80. — P. L947-954.
10. Neutrophils, not complement, mediate the mortality of experimental hemorrhagic pancreatitis /C. Kyriakides, J. Jasteen, Y. Wang et al. //Pancreas. — 2001. — Vol. 22. — P. 40-46.
11. Occurrence of the adult respiratory distress syndrome in neutropenic patients /R.J. Maundler, R.C. Hackman, E. Riff et al. //Am. Rev. Respir. Dis. — 1986. — Vol. 133. — P. 313-316.
12. Organism-specific neutrophil-endothelial cell interactions in response to Esherichia coli, Streptococcus pneumoniae and Staphylococcus aureus /J.G. Moreland, G. Bailey, W.M. Nauseef, J.P. Weiss //J. Immunol. — 2004. — Vol. 172. — P. 426-432.
13. The effects of leucodepletion in patients who develop the systemic inflammatory response syndrome following cardiopulmonary bypass /D.F. Treacher, M. Sabbato, K.A. Brown, V.A. Gant //Perfusion.
— 2001. — Vol. 16. — P. S67-74.
14. Time-course of sTREM (soluble triggering receptor expressing on myeloid cells)-1, procalcitonin, and C-reacteve protein plasma concentration during sepsis /S. Gibot, A.A. Cravoisy, M.-N. Kolopp-Sarda et al. //Crit. Care Med. — 2005. — Vol. 33. — P. 792-796.
15. Upregulation of reactive oxygen species generation and phagocytosis and an increased apoptosis in human neutrophils during severe sepsis and septic shock /P.S. Martins, E.G. Kalla, M.C. Neto et al. //Shok. — 2003. — Vol. 20. — P. 208-212.
16. Wagner, J.G. Neutrophil migration with an emphasis on the pulmonary vacculature /J.G. Wagner, R.A. Roth //Pharmacol. Rev. — 2000.
— Vol. 52. — P. 349-374.
17. Windsor, A.C.J. Role of the neutrophil in adult respiratory distress syndrome /A.C.J. Windsor //Br. J. Surg. — 1993. — Vol. 80. — P. 10-17.
■ ■ 4
ПОЛИТРАВМА
Клинические исследование кровоизлияние: Дексразоксан — Реестр клинических исследований
Случайная экстравазация антрациклинов, например доксорубицин и его производное эпирубицин, может вызывать прогрессирующее разрушение тканей, включая серьезные повреждения кожи, подкожная ткань, мышцы и нервы.
Пациент может страдать от острых местных симптомов, таких как боль и отек, которые могут прогрессировать в образование пузырей и некроз. Впоследствии дизестезия, атрофия кожи, обезображивание, следствием этого может быть нарушение функции конечностей.
Требуется хирургическое удаление всех пораженных тканей, и часто требуется хирургическая обработка раны. расслоение кожи. Таким образом, пациент подвергается тяжелой хирургической операции, которая в очередь приводит к отсрочке дальнейшего цитотоксического лечения.
Доклинические исследования на животных, а также клинические многоцентровые исследования фазы II продемонстрировали: очень значимая эффективность дексразоксана в предотвращении разрушения тканей, вызванного антрациклинов.
Это подтверждающее исследование определит действие Топотекта® (дексразоксана) как острого антидот у пациентов с экстравазацией антрациклина.
Статус орфанного лекарства TopoTarget A / S был признан Topotect® лекарством для сирот продукт для лечения экстравазации антрациклина Европейской Комиссией в Сентябрь 2001 г. и FDA в ???.
Цель
Основные задачи:
• Чтобы предотвратить прогрессирование экстравазации антрациклинового поражения в виде язвы и некроз, требующий хирургического вмешательства
Вторичные цели:
— Предотвратить развитие некроза и разрушения глубоких тканей, приводящих к поздним последствиям как нарушение функции конечностей и неврологический дефицит
— Чтобы предотвратить отсрочку запланированного лечения рака из-за лечения кровоизлияние
— Чтобы оценить переносимость и / или токсичность Топотекта®, используемого по этому показанию, по указанному расписанию
Дизайн исследования Это открытое нерандомизированное исследование фазы II / III. Тридцать пять оцениваемых пациенты с экстравазией антрациклина получают лечение.
Экстравазация определяется наличием боли и / или отека и / или покраснения в области место, где предположительно произошла утечка антрациклина. Экстравазии впоследствии подтверждается у каждого пациента с помощью флуоресцентной микроскопии не менее двух точек биопсии во время аварии.
Критерии успеха Предупреждение хирургического вмешательства, некроза и отдаленных последствий оценивается через 3 месяца после экстравазации.
Функции безопасности
— Токсичность, вызванная Topotect®, будет проверена гематологическим и химическим анализом крови, вопросы задаются по поводу любого дискомфорта. Плановые клинические осмотры проводятся
— Систематическая клиническая оценка отмеченного участка кожи, покрывающего область экстравазация будет выполнена, чтобы оценить необходимость операции
— Будут сделаны последовательные цветные фотографии пораженной кожи.
Медицинское лечение Пациентам вводят внутривенное вливание Топотекта®. один раз в день три дня подряд в следующих дозах: 1000 мг / м2 + 1000 мг / м2 + 500 мг / м2. Первая доза вводится как можно скорее и в течение 6 часов после экстравазация и следующие две дозы через 24 и 48 часов после первой инфузии.
Патогенез индуцированного гриппом острого респираторного дистресс-синдрома
Kirsty R Short, Edwin J B Veldhuis Kroeze, Ron A M Fouchier, Thijs Kuiken Острый респираторный дистресс-синдром (ОРДС) — это летальное осложнение гриппа. В этом обзоре мы предлагаем комплексную модель его патогенеза. ОРДС включает в себя: повреждение эпителиального и эндотелиального компонентов аэро-гематического барьера легких (здесь и далее будет использоваться аббревиатура “АГБ”, под которой будет подразумеваться его эпителиальный и эндотелиальный компоненты — прим. редактора), просачивание жидкости в просвет альвеол, а также дыхательную недостаточность. Самая важная часть АГБ — это альвеолярный эпителий, клетки которого связаны плотными контактами. Именно эти клетки являются мишенью вируса гриппа, который подавляет в них активность натрий-калиевого насоса, повреждает плотные соединения, и в конечном счете приводит к гибели инфицированных клеток. Зараженные эпителиальные клетки продуцируют цитокины, которые привлекают лейкоциты — нейтрофилы и макрофаги — и активируют соседние эндотелиальные клетки. Активированные эндотелиальные клетки и проникшие лейкоциты стимулируют дальнейшую инфильтрацию, вместе с этим лейкоциты индуцируют продукцию активных форм кислорода и оксида азота, которые повреждают барьер. Активированные макрофаги также вызывают апоптоз эпителиальных клеток. Модель индуцированного гриппом ОРДС отлична от классической модели, центром которой является повреждение эндотелия, и приводит обоснования для терапевтического вмешательства с целью смягчения ответа организма при индуцированном вирусом гриппа ОРДС.Введение
Вирус гриппа А — это респираторный патоген, который существенно влияет на здоровье человека во всем мире. В человеческой популяции циркулируют сезонные вирусы, которые вызывают ежегодные эпидемии, уносящие приблизительно 500000 жизней каждый год. Новый штамм вируса гриппа А, к которому популяция еще не приобрела иммунитет, может стать причиной глобальной пандемии с изменчивой смертностью; пандемический h2N1 2009 года вызвал 151700-575400 смертельных случаев в первый год циркуляции, в то время, как пандемический h2N1 1918 года привел более чем к 40 миллионам смертей. Существует опасение, что новые возникающие из животных-резервуаров штаммы вируса гриппа А (например, происходящие от птиц вирусы H5N1 и H7N9) могут приобрести высокую контагиозность у людей и вызвать серьезную пандемию. Чтобы смягчить тяжесть такой пандемии, следует понимать механизмы, путем которых грипп А вызывает респираторное заболевание. Основное осложнение инфицирования вирусом гриппа — вирусная пневмония, которая часто происходит совместно или с последующей бактериальной пневмонией. В этом обзоре мы фокусируемся на первичной вирусной пневмонии, которая может привести к острому респираторному дистресс-синдрому (ОРДС). Клинически острая фаза ОРДС характеризуется цианозом, гипоксемией, отеком легких и нарастающей в течение долгого времени дыхательной недостаточностью, которые приводят к полиорганной недостаточности и высокой смертности (до 60%). Помимо вируса гриппа А к ОРДС могут привести другие нарушения, к примеру — сепсис, пневмония, травма и аспирация желудочного содержимого. Главной причиной дыхательной недостаточности в острую фазу ОРДС является повреждение АГБ, который обеспечивает газообмен (рисунок 1). Повреждение этого барьера приводит к заполнению просвета альвеол богатым белком экссудатом, который также содержит фибрин, эритроциты и воспалительные клетки. Эта отечная жидкость уменьшает альвеолярный газообмен и может приводить к тяжелой дыхательной недостаточности, как это было отмечено у пациентов с ОРДС. Понимание механизмов повреждения АГБ в острую фазу ОРДС достигнуто в основном благодаря исследованиям бактериального сепсиса, при котором в первую очередь повреждается эндотелий. Эта модель может не подходить для вируса гриппа, который первым инфицирует эпителий. Кроме того, хотя некоторые обзоры фокусируются на различных факторах индуцированного вирусом гриппа повреждения легких (включая факторы вирулентности вируса, продукцию цитокинов и патологические изменения), нет такого обзора, который бы рассматривал все эти факторы в совокупности. Мы рассматриваем имеющуюся литературу о взаимодействии между вирусом гриппа А и ключевыми типами клеток, представленными в легочных альвеолах, в острой фазе ОРДС. В частности, мы обращаем особое внимание на роли эпителиальных и эндотелиальных клеток легких, нейтрофилов и макрофагов в повреждении АГБ, разрабатывая новую концептуальную основу для индуцированного гриппом ОРДС.Эпителиальные клетки
В числе первых клеток, которые встречает вирус гриппа после входа в альвеолы — эпителиальные клетки, они же пневмоциты 1 или 2 типов. Пневмоциты 1 типа — это плоские клетки, покрывающие 95% поверхности альвеол и обеспечивающие легкую диффузию газа между воздухом в просвете альвеол и кровью в капиллярах. Пневмоциты 2 типа — кубические клетки, секретирующие легочный сурфактант, который важен для снижения альвеолярного поверхностного натяжения. Плотные контакты между эпителиальными клетками отвечают за снижение проницаемости эпителиального слоя альвеолярных клеток. Плотные контакты образованы различными белками, включающими клаудины и запирающие зоны 1, 2 и 3. Более 90% сопротивления транспорту белков через АГБ обусловлено альвеолярным эпителием. Ограничивая транспорт белка, эпителий поддерживает осмотическое давление по разные стороны барьера и предотвращает отек легких. Второй механизм, с помощью которого альвеолярный эпителий предотвращает экссудацию жидкости в просвет альвеол, заключается во взаимодействии ионных каналов и мембранных белков, к которым относятся эпителиальные натриевые каналы (ENaCs), трансмембранный регулятор проводимости муковисцидоза и много различных аквапоринов. Наиболее хорошо описанными ионными каналами в легких являются ENaCs, которые присутствуют на апикальной поверхности пневмоцитов обоих типов. Этот канал дополняется Na+/K+ АТФазой на базолатеральной поверхности клетки. Ионы Na+, входящие в канал на апикальной поверхности клетки, перемещаются к базолатеральной поверхности, где Na+/K+ АТФаза выкачивает их в подлежащий интерстиций. Наличие Na+ в интерстиции создает осмотический градиент, который удаляет воду из просвета альвеол через аквапорины и внутриклеточные пути в альвеолярных эпителиальных клетках, тем самым предотвращая отек легких. Пневмоциты являются не только важнейшим компонентом эпителиально-эндотелиального барьера, но также и клетками-мишенями вируса гриппа А. Вирус гриппа А связывается с клеткой-мишенью путем прикрепления вирусного гемагглютинина к сиалосахариду на поверхности клетки. Экспрессия сиалосахарида различна для различных пневмоцитов: пневмоциты 1 типа в основном экспрессируют α-2,6-связанные сиалосахариды, которые “предпочитают” вирусы гриппа человека, в то время как пневмоциты 2 типа экспрессируют в основном α-2,3-сцепленные сиалосахариды, которые “предпочитают” птичьи вирусы гриппа. Результаты вскрытий пациентов, погибших от гриппа H5N1, показали, что антигены вируса гриппа H5N1 наиболее часто встречались в пневмоцитах второго типа, что согласуется с данными о тропизме птичьих вирусов гриппа к α-2,3-сцепленным сиалосахаридам. Напротив, результаты вскрытия погибших в результате заражения пандемическим вирусом h2N1 2009 года (который имеет двойную тропность, к α-2,3-сцепленным и α-2,6-сцепленным сиалосахаридам) показали экспрессию антигенов пандемического вируса h2N1 в обоих типах пневмоцитов. Эти вирус-зависимые различия в прикреплении зависят от специфических аминокислотных остатков в рецептор-связывающем домене глобулярной головки гемагглютинина. Позиции 190 и 225 (вирус гриппа А Н1) и позиции 226 и 228 (вирусы гриппа А Н2 и Н3) являются основными детерминантами рецепторной специфичности пандемических вирусов гриппа. Мутации в этих позициях также определяют рецептор-связывающие свойства вируса гриппа А H5N1. Таким образом, тип клеток-мишеней альвеолярного эпителия может различаться у разных штаммов гриппа А. Источник: журнал Lancet (А) В нижних дыхательных путях трахея разделяется на главные бронхи и несколько уровней бронхов и бронхиол, оканчиваясь терминальными бронхиолами. (В) Каждая терминальная бронхиола содержит в ацинусе около 2000 альвеол. В стенках альвеол содержится сеть легочных капилляров, подающих кровь, приходящую из легочных артериол и дренируемую легочными венами. (С) Каждая альвеола содержит несколько альвеолярных макрофагов и выстлана плоскими пневмоцитами 1 типа и кубическими пневмоцитами 2 типа. Пневмоциты лежат на базальной мембране, которая непосредственно примыкает или сливается с базальной мембраной легочных капилляров, которые выстланы эндотелиальными клетками. (D) Газообмен между кровью в легочных капиллярах и воздухом в альвеолярном просвете происходит через АГБ, состоящий из альвеолярного эпителиального слоя, базальной мембраны или мембран и легочного эндотелиального слоя (Е) Специфическое окрашивание на панкератин пневмоцитов 1 типа (стрелки в виде наконечника) и пневмоцитов 2 типа (обычные стрелки) в нормальных человеческих альвеолах (уменьшено на 13% от ×1000). (F) Специфическое окрашивание на фактор фон Виллебранда эндотелиальных клеток капилляров (стрелка) в последовательном сечении ткани, показанной в разделе Е (уменьшено на 13% от ×1000). БМ = базальная мембрана. ПК = плотный контакт. Вскоре после инфицирования вирус гриппа А вызывает накопление жидкости в просвете альвеол путем прямого ингибирования ENaCs (рисунок 2). Экспозиция пневмоцитов типа 2 in-vitro по вирусу гриппа А/Пуэрто Рико/8/1934 (h2N1) в течение 1 часа уменьшила активность ENaC при сохранении целостности эпителия. Это уменьшение активности было связано с активацией протеинкиназы С и фосфолипазы вирусным гемагглютинином. Ионный канал вируса гриппа М2 также может ингибировать активность ENaCs на эпителиальных клетках, вызывая продукцию активных форм кислорода и последующую активацию протеинкиназы С. Крысы, инфицированные вирусом гриппа и затем получившие интратрахеально физиологический раствор, имели достоверно меньший клиренс жидкости из легких через два часа после заражения, чем неинфицированные крысы, что согласуется с вышеизложенными данными. На более поздней стадии инфекции наиболее важную роль в повреждении АГБ путем разрушения эпителиального монослоя играет индуцированная вирусом гриппа А гибель эпителиальных клеток. В тканях пациентов, погибших от вызванного вирусом гриппа ОРДС наблюдались оба типа клеточной смерти — как апоптоз, так и некроз. Механизм, посредством которого вирус гриппа индуцирует клеточную смерть, был показан in vitro, хотя он типичнее для бронхиальных или бронхиолярных эпителиальных клеток, чем для альвеолярных эпителиальных клеток. Инфицирование линий человеческих бронхиолярных эпителиальных клеток (NCL-h392) рекомбинантным вирусом гриппа А/Англия/939/69 (h4N2) привело к каспаза-8-зависимому апоптозу. Вирус гриппа может индуцировать апоптоз эпителиальных клеток посредством разнообразных путей, включая активацию протеинкиназы R, которая впоследствии активирует проапоптозные гены (нап. FAS). Кроме того, вирусная нейраминидаза может приводить трансформирующий фактор роста β в его активную форму, которая затем индуцирует апоптоз путем взаимодействия с соответствующими рецепторами. Экспрессия неструктурного белка 1 (NS-1), полученного из вируса гриппа А/Гонконг/483/97 (H5N1), также достаточна, чтобы вызвать каспаза-зависимый апоптоз эпителиальных клеток. В отличие от индуцированного вирусом гриппа апоптоза клеток NCL-h392, заражение эмбриональных человеческих бронхиальных эпителиальных клеток адаптированным к мышам штаммом вируса гриппа А/Пуэрто Рико/8/1934 (h2N1) привело к некрозу. Вирусный полимеразный комплекс (в частности нуклеопротеин) может способствовать индуцированной вирусом гриппа клеточной смерти путем удаления кэп-структур мРНК клетки-хозяина (так называемый “захват кэпа”), что уменьшает количество кэпированной мРНК хозяина, которая может транслироваться с получением функциональных белков. Из-за важной роли эпителия в минимизации отека легких, утрата эпителиальных клеток в результате индуцированной вирусом гриппа клеточной смерти играет важную роль в повреждении структуры легких. Источник: журнал Lancet На ранних стадиях ОРДС, вирус гриппа А может ингибировать активность ENaCs (i) на альвеолярных эпителиальных клетках. Позднее вирус гриппа может вызывать апоптоз или некроз (ii) клеток альвеолярного эпителия, продукцию цитокинов (iii) и нарушение плотных контактов (iv). В конечном счете, эти изменения приводят к накоплению белоксодержащей жидкости в альвеолах (розовый). Специфическое окрашивание на нуклеопротеин гриппа А демонстрирует заражение гриппом А H5N1 (вставка) клеток альвеолярного эпителия (стрелка) легочной ткани человека через 24 часа (уменьшено на 22% от ×400). (B) После заражения вирусом гриппа А, эндотелиальные клетки капилляров позволяют экстравазацию моноцитов (v) и нейтрофилов (vi) в альвеолы посредством активации молекул адгезии, таких как Е-селектин и Р-селектин. На вставке показана адгезия нейтрофилов (стрелка) и других лейкоцитов к эндотелию кровеносного сосуда легких хорька, через 4 дня после заражения вирусом гриппа Н5N1 in-vivo (окрашивание гематоксилин-эозином, уменьшено на 22% от ×800). Эндотелий может продуцировать различные провоспалительные цитокины (vii) в ответ на грипп А. В обоих (А) и (В) эпителиальных клетках представлены оба типа пневмоцитов. ОРДС = острый респираторный дистресс синдром. БМ = базальная мембрана. Эр = эритроцит. ПК = плотный контакт. TNF = фактор некроза опухоли. Воздействие гибели эпителиальных клеток на повреждение АГБ может быть усугублено способностью вируса гриппа повреждать плотные контакты между эпителиальными клетками. В частности, некоторые штаммы H5N1 могут непосредственно разрушать плотные контакты через представленный в консенсусной последовательности ESEV на карбоксильном конце вирусного NS-1 мотив, связывающий PDZ-лиганд. Эта последовательность опосредует прикрепление вирусного NS-1 к белкам хозяина, таким как scribble и DLG1, которые играют важную роль в формировании плотных контактов. Соответственно, заражение клеток собачьей почки Madin-Darby вирусом, обладающим мотивом ESEV, нарушает образование плотных контактов. Тем не менее, мотив ESEV экспрессируется не всеми штаммами вируса гриппа А. Таким образом, прямое нацеливание вирусного NS-1 на плотные контакты происходит не во всех случаях заражения вирусом гриппа. Наконец, зараженные вирусом гриппа эпителиальные клетки продуцируют цитокины, которые могут впоследствии повредить АГБ. Посмертный анализ летального случая пневмонии, вызванной вирусом гриппа H5N1, показал продукцию фактора некроза опухоли α (TNFα) альвеолярными эпителиальными клетками. По данным исследования in-vitro, инфицированные вирусом гриппа альвеолярные эпителиальные клетки продуцируют и другие цитокины помимо TNFα (таблица 1). Как правило, к данным исследований in-vitro необходимо относиться с осторожностью, так как результаты могут меняться в зависимости от штамма вируса гриппа и типа используемых клеток, а также потому, что во многих исследованиях используются эмбриональные клетки или клеточные линии, полученные из трахеи, бронхов или бронхиол (вместо альвеолярных). По результатам исследований на зародышевых человеческих пневмоцитах 2 типа можно предположить, что увеличение продукции CXCL10, интерлейкина 6 и CCL5 после заражения H5N1 (по сравнению с заражением сезонным штаммом h2N1) может способствовать увеличению тяжести заболевания. Однако механизм, посредством которого те или иные цитокины повреждают АГБ, остается неясным. Интерлейкин 1β и подобные ему цитокины могут подавлять активность ENaC. Плотные контакты могут разрушаться под действием таких цитокинов, как TNFα. Кроме того, некоторые цитокины, продуцируемые альвеолярными эпителиальными клетками в ответ на заражение вирусом гриппа, функционируют как хемоаттрактанты, либо могут повышать экспрессию молекул клеточной адгезии на эндотелиальных клетках легких и, таким образом, обеспечивать выход лейкоцитов из сосудов. Впоследствии эти клетки могут повреждать АГБ. Таблица 1. Продукция цитокинов различными типа клеток после заражения эпителиальных клеток вирусом гриппа А.*Определяется экспрессией мРНК. TNF = фактор некроза опухоли.
Эндотелиальные клетки
Эндотелиальные клетки — наиболее распространенный тип клеток в легких, составляющий 30% от общего количества клеток. В альвеолах эндотелиальные клетки покрывают изнутри капилляры, которые формируют сеть в стенках альвеол. Своей апикальной стороной эндотелиальные клетки находятся в непосредственном контакте с циркулирующей кровью и формируют участок прикрепления для рекрутированных воспалительных клеток. После соответствующей активации, эндотелиальные клетки экспрессируют молекулы клеточной адгезии, которые связываются с родственными им лигандами на лейкоцитах и опосредуют выход лейкоцитов из сосудов. Своей базолатеральной стороной эндотелиальные клетки лежат на базальной мембране, которая тесно соединена (или даже сливается) с эпителиальной базальной мембраной. Это тесное взаимодействие предполагает, что эндотелиальные клетки подвержены сильному влиянию сигналов и вирусных частиц, высвобождаемых из эпителиальных и воспалительных клеток в просвет альвеол. Активация эндотелия этими сигналами и вирусными частицами — необходимое звено эффективного иммунного ответа против гриппа А. Однако в то же время активация эндотелия может играть роль в повреждении АГБ и способствовать отёку легких. Заражение вирусом гриппа может активировать экспрессию молекул адгезии эндотелиальных клеток (к примеру — Е-селектин, Р-селектин, ICAM1 и VCAM1), тем самым обеспечивая проникновение лейкоцитов в альвеолы (рисунок 2). Присутствие большого количества нейтрофилов и макрофагов может повреждать АГБ посредством различных механизмов. Заражение вирусом гриппа А H5N1 приводит к более высокой экспрессии Е-селектина и Р-селектина на эндотелиальных клетках человека, нежели чем заражение пандемическим h2N1 1918 года или сезонным штаммом h2N1. Эта разница в активации эндотелия может помочь объяснить увеличение привлечения лейкоцитов, которое происходит при заражении вирусом гриппа H5N1 и сопровождается возрастающим повреждением альвеол. Заражение вирусом гриппа может также вызывать продукцию цитокинов эндотелиальными клетками легких. Сезонные вирусы h4N2 и h2N1 вызывают продукцию интерлейкина 6, CXCL9 и CXCL 10 эндотелиальными клетками пупочной вены человека, в то время как заражение вирусом H5N1 может вызвать продукцию CXCL10, TNFα и интерлейкина 6. В исследованиях на мышах было показано, что индуцированная вирусом гриппа активация легочного эндотелия увеличивает тяжесть заболевания у мышей, инфицированных пандемическим вирусом h2N1 2009 года или сезонным вирусом h2N1. В частности, легочный эндотелий продуцировал цитокины, такие как CCL2, CXCL10, интерферон α, TNFα и интерферон γ в ответ на вирус гриппа А и рекрутировал лейкоциты в легкие в течение инфекции. Соответственно, ингибирование продукции цитокинов эндотелием и накопления лейкоцитов в результате лечения агонистом сфингозин-1-фосфата значительно улучшило выживаемость после инфицирования вирусом гриппа. К сожалению, механизм, посредством которого цитокины эндотелиального происхождения могут повреждать АГБ при заражении вирусом гриппа, остается неизвестен. Тем не менее, эти данные подчеркивают важную роль, которую играет активация эндотелия в индуцированном вирусом гриппа повреждении альвеол. Вирус гриппа А может также повреждать АГБ, заражая непосредственно эндотелиальные клетки, вызывая их гибель и, таким образом, создавая достаточное для развития отека легких повреждение эндотелия. In vitro наличие многоосновного участка рестрикции в геммаглютинине позволяет вирусу H5N1 продуктивно реплицироваться в эндотелиальных клетках. У домашних птиц геммаглютинины с многоосновным участком рестрикции обеспечивают системную репликацию вируса, поскольку эти геммаглютинины могут быть расщеплены экспрессируемыми повсеместно протеазами, в отличие от геммаглютининов с одноосновными участками рестрикции, которые зависят от трипсиноподобных протеаз, экспрессируемых только в дыхательных путях и желудочно-кишечном тракте. Однако эндотелиальный слой играет не такую важную роль в предотвращении выхода жидкости из просвета сосуда в просвет альвеолы, как эпителиальный слой. Вместе с базальной мембраной эндотелиальный слой обеспечивает лишь 10% от сопротивления к транспорту белка (который поддерживает осмотическое давление) через АГБ. Таким образом, изолированное повреждение эндотелиального слоя не является ключевой причиной отека легких.Нейтрофилы
Нейтрофилы прибывают в легочные альвеолы в течение первого дня заражения вирусом гриппа, где они присоединяются к резидентным лейкоцитам, таким как альвеолярные макрофаги. Нейтрофилы — недолго живущие фагоцитирующие гранулоциты, которые в ответ на провоспалительную стимуляцию просачиваются через стенку сосуда из крови в просвет альвеолы. Экстравазация нейтрофилов происходит в несколько шагов: роллинг по эндотелию, сцепление с адгеринами и селектинами эндотелиальных клеток, миграция через эпителиальный и эндотелиальный слои в альвеолярный просвет. Эта миграция может вызвать временное, но недостаточное для развития отека легких, повреждение АГБ. После попадания в просвет альвеолы нейтрофилы активируются в ответ на местно представленные цитокины и патогены. Активированные нейтрофилы могут фагоцитировать патогены. В результате, фагосомы могут сливаться с цитоплазматическими первичными, вторичными и третичными гранулами, которые содержат токсичные соединения, уничтожающие фагоцитированные патогены. Эти токсичные соединения могут быть выбрасываться наружу после контакта с неперевариваемым материалом (например, иммунными комплексами, осажденными на базальной мембране), фагоцитоза мембранолитических веществ (например, кристаллов уратов) или слияния гранул с фагосомой прежде, чем она полностью закроется. В дополнение к фагоцитозу, нейтрофилы могут ловить и убивать патогены, формируя нейтрофильные внеклеточные ловушки (NETs), которые состоят из внеклеточной ДНК, обсемененной гистонами, хроматином и антимикробными соединениями. Важность нейтрофилов в ограничении репликации и распространения вируса гриппа отражает тот факт, что мыши с дефицитом нейтрофилов имели высокие титры вируса в легких и других органах и тканях в сравнении с контрольными мышами. Тем не менее, нейтрофилы могут также играть важную роль в повреждении АГБ. В случаях ОРДС у человека, концентрации нейтрофилов в жидкости бронхоальвеолярного лаважа положительно коррелировали с тяжестью заболевания. Подобным образом, повреждение легких у зараженных вирусом гриппа мышей снизилось при блокировке накопления нейтрофилов в легких. Хотя было предложено несколько механизмов, путем которых нейтрофилы могут повреждать АГБ при ОРДС, лучше всего описаны механизмы, основанные на способности нейтрофилов продуцировать активные формы кислорода, цитокины и выбрасывать NETs (рисунок 3). Нейтрофилы могут продуцировать активные формы кислорода (например супероксид) посредством НАДФ-оксидазы, многокомпонентного ферментативного комплекса, состоящего из белков в цитозоле и цитохрома b558 на мембране вторичных гранул. Результатом слияния вторичных гранул с фагосомами или клеточной мембраной является продукция активных форм кислорода в фагосоме или внеклеточной среде. Соответственно, активные формы кислорода могут не только уничтожать патогены, такие как вирус гриппа, но также и повреждать альвеолы. В соответствии с этим представлением, устранение супероксида (путем инъекции супероксид дисмутазы) предотвращает летальный исход инфекции гриппа у мышей. Точно также заражение вирусом гриппа вызывает меньшие повреждения тканей у дефицитных по НАДФ-оксидазе мышей, чем у мышей дикого типа. Источник: журнал Lancet (А) Нейтрофилы могут повредить аэро-гематический барьер (АГБ) при заражении вирусом гриппа посредством продукции активных форм кислорода (i). В ответ на заражение вирусом гриппа нейтрофилы также продуцируют цитокины (ii), которые могут затем опосредованно повредить АГБ. Производство NETs (iii) может также повредить эпителиальные и эндотелиальные клетки. На вставке показаны NETs (стрелка) в крупных кровеносных сосудах мыши после заражения вирусом гриппа in-vivo (окрашивание гематоксилин-эозином). (В) Макрофаги могут повредить АГБ посредством индуцированной гриппом экспрессии TRAIL, который взаимодействует с DR5 (iv), вызывая апоптоз эпителиальных клеток. Экспрессия TRAIL инициируется интерфероном β. В ответ на вирус гриппа макрофаги также продуцируют различные цитокины (v), которые могут косвенно повредить АГБ. На вставке показаны макрофаги (стрелка) взаимодействующие с эпителиальными клетками альвеол легочной ткани человека через 24 часа после заражения ex-vivo вирусом гриппа A H5N1 (окрашивание гематоксилин-эозином, уменьшено на 33% от ×500). В конечном результате грипп А индуцирует опосредованную макрофагами продукцию NO (vi) и последующее образование пероксинитрита, которые могут повредить АГБ. В обоих (А) и (В) эпителиальных клетках представлены оба типа пневмоцитов. Вновь рекрутированные макрофаги изображены темно-розовыми, резидентные альвеолярные макрофаги изображены зеленым. Накопление экссудата в альвеолах показано розовым. ОРДС = острый респираторный дистресс синдром. БМ = базальная мембрана. АФК = активные формы кислорода. НВЛ = нейтрофильная внеклеточная ловушка. ФНО = фактор некроза опухоли. NO = оксид азота. DR5 = рецептор смерти 5. TRAIL = фактор некроза опухоли связанный с апоптоз-индуцирующим лигандом. Вставка части iii воспроизведена Narasaraju и коллегами с разрешения Elsevier. Миелопероксидаза представлена в первичных гранулах и создает условия для продукции хлорноватистой кислоты из перекиси водорода и свободных ионов хлора. Взаимодействуя с маннозными рецепторами макрофагов, миелопероксидаза может спровоцировать высвобождение макрофагами активных форм кислорода и цитокинов. Эти эффекты могут не только инактивировать вирус гриппа, но и вызвать повреждение АГБ. Соответственно, заражение вирусом гриппа в меньшей степени приводит к разрушению легочной структуры и отеку легких у миелопероксидаза-дефицитных мышей, чем у мышей дикого типа. Кроме того, миелопероксидаза-дефицитные мыши также демонстрируют повышенную экспрессию клаудина 9 и клаудина 18-1. Эти данные позволяют предположить, что миелопероксидаза может привести к повреждению АГБ путем разрушения плотных контактов. Все еще не выяснено, какую роль индуцированное миелопероксидазой повреждение играет в формировании смертности по причине инфекции вирусом гриппа. Хотя выживаемость мышей дикого типа не отличалась от таковой у зараженных большой дозой вируса Mpo -/- мышей, у дефицитных мышей имелась тенденция к длительному выживанию при более низкой дозе. Нейтрофилы могут опосредованно повреждать АГБ путем продукции цитокинов. Инфицирование вирусом гриппа побуждает нейтрофилы к продукции CXCL2 и интерлейкина 8, которые рекрутируют дополнительные нейтрофилы к инфицированной области. Также после заражения вирусом гриппа нейтрофилы становятся основными продуцентами CXCL10. Инфицирование вирусом гриппа вызывает меньшие легочные повреждения и смертность у CXCL10-дефицитных мышей, чем у мышей дикого типа. Выделяемый нейтрофилами CXCL10 может действовать аутокринным образом и связываться с рецепторами CXCR3, которые экспрессируются на нейтрофилах. Этим путем CXCL10 индуцирует генерацию супероксида и усиливает хемотаксис по направлению к CXCL2. В соответствии с этим, лечение хорьков антагонистом Cxcr3 снижает тяжесть клинических проявлений вирусной инфекции H5N1. Нейтрофилы также могут повреждать АГБ за счет производства NETs. У мышей, инфицированных вирусом гриппа А/Пуэрто Рико/8/1934, нейтрофильные внеклеточные ловушки формировались и прикреплялись к эндотелию в областях кровоизлияний, как в просвете альвеол, так и в терминальных бронхиолах в областях повреждения тканей. Эти результаты свидетельствуют о том, что NETs могут повреждать как эпителиальные, так и эндотелиальные клетки альвеол. Роль вируса гриппа в продукции нейтрофильных внеклеточных ловушек in vivo была подтверждена экспериментами in vitro, путем ко-инкубации нейтрофилов и инфицированных вирусом гриппа эпителиальных клеток. Однако выживаемость мышей, дефицитных по пептидиларгининдезиминазе 4 (ферменту, который необходим для формирования NETs) после заражения вирусом гриппа в основном не отличается от мышей дикого типа; предполагается, что формирование нейтрофильных внеклеточных ловушек может не быть критичным для повреждения АГБ.Макрофаги
Как и нейтрофилы, макрофаги — это важный компонент врожденного иммунного ответа, направленного против вируса гриппа. Макрофаги поглощают патогены и инфицированные, либо апоптотические клетки. Фагоцитированные патогены затем уничтожаются посредством респираторного взрыва и продукции активных форм кислорода, либо оксида азота. Макрофаги могут также производить широкий спектр как провоспалительных, так и противовоспалительных цитокинов. В пределах альвеол следует различать два типа макрофагов. Первый тип — альвеолярные макрофаги, которые являются длительно живущими резидентными клетками, которые встречаются до 7 на альвеолу. Они, как правило, несут альтернативно активируемый фенотип, минимизирующий воспаление легких в ответ на безвредные патогены, защищая при этом легкое от более вирулентных возбудителей. Их значение в контроле за инфицированием вирусом гриппа подтверждается тем фактом, что удаление альвеолярных макрофагов до инфицирования вирусом гриппа приводит к повышению вирусной репликации и утяжелению заболевания. При вызванной вирусом гриппа инфекции моноциты, привлекаемые в альвеолы хемокинами (например, CCL2, CCL5 и CXCL10), вскоре значительно превосходят альвеолярные макрофаги в количестве. Эти моноциты мигрируют из крови в просвет альвеол, где они дифференцируются в макрофаги. Этот второй тип макрофагов — недавно рекрутированные макрофаги — обычно несет классически активированный фенотип. Хотя блокировка пополнения рядов моноцитов в легком приводит к увеличению репликации вируса, недавно рекрутированные макрофаги также могут играть важную роль в повреждении АГБ. После инфицирования вирусом гриппа, у мышей с дефицитом CCR2 (рецептора для CCL2, который опосредует хемотаксис моноцитов) уменьшались как привлечение моноцитов в легкие, так и тяжесть легочных поражений. Основные механизмы повреждения недавно рекрутированными макрофагами связаны с высвобождением апоптоз-индуцирующего лиганда, связанного с фактором нероза опухоли (TRAIL, также известного как TNFSF10), продукцией оксида азота и провоспалительных цитокинов (рисунок 3). После заражения вирусом гриппа А/Пуэрто-Рико/8/1934 недавно рекрутированные макрофаги экспрессировали TRAIL, который взаимодействует с рецептором смерти 5, белком, который активируется на альвеолярных эпителиальных клетках инфицированных вирусом гриппа; такое взаимодействие индуцировало апоптоз этих клеток. Соответственно, ингибирование TRAIL снижало скорость апоптоза эпителиальных клеток, степень отека легких и смертность мышей, инфицированных вирусом гриппа А. Продукция TRAIL зависит от активации вирусом гриппа А протеинкиназы R и последующего производства интерферона β. Аутокринное взаимодействие интерферона β со своим рецептором (рецептором интерферонов α, β, и ω) вызывает производство TRAIL JAK–STAT-зависимым образом. В соответствии с этим путем, блокировка сигнала интерферона β у мышей нарушает продукцию TRAIL и снижает повреждение альвеолярного эпителия после инфицирования вирусом гриппа. Макрофаги и, в меньшей степени, нейтрофилы, которые стимулируются провоспалительными цитокинами, экспрессируют индуцибельную NO-синтазу (NOS2), которая опосредует образование оксида азота. Оксид азота может комбинироваться с супероксидом, формируя пероксинитрит, который может как убивать патогены, так и повреждать клетки. Ингибирование активности NOS2 сокращает масштабы пневмонии и смертность после летальной инфекции вирусом гриппа. Эти данные согласуются с данными о том, что, в отличие от мышей дикого типа, у NOS2-дефицитных мышей после инфицирования вирусом гриппа не развивалась пневмония. Karupiah и коллеги предположили, что экспрессия NOS2 вносит больший вклад в тяжесть пневмонии, чем в цитопатический эффект вируса гриппа, поскольку увеличение титров вируса у NOS2-дефицитных мышей путем блокирования интерферона γ все же не вызывает летального исхода. Результаты этого исследования подчеркивают важность повреждения легких, индуцированного оксидом азота при заражении вирусом гриппа. Макрофаги могут продуцировать широкий спектр цитокинов в зависимости от штамма вируса гриппа и типа макрофага. Важность этих цитокинов для защиты от вирусов гриппа доказана тем фактом, что вирусные NS-1 действуют на нескольких уровнях, служа препятствием продукции интерферонов и интерферон-опосредованной индукции противовирусных белков. К примеру, мутация Asp92Glu в NS-1 превращает вирус гриппа А H5N1 в относительно нечувствительный к интерферону α, интерферону γ и TNFα. Однако, как правило, заражение макрофагов вирусом H5N1 приводит к синтезу более разнообразного репертуара цитокинов в более высоких концентрациях, чем при заражении другими штаммами вируса гриппа А. Несмотря на то, что эта индукция является важным компонентом противовирусного ответа, она также может способствовать гиперцитокинемии и последующей легочной дисфункции, отмеченной в некоторых случаях заболевания людей вирусом гриппа H5N1. Человеческие макрофаги моноцитарного происхождения, возможные представители недавно рекрутированных макрофагов, производят значительно более высокие концентрации провоспалительных цитокинов ,чем человеческие альвеолярные макрофаги при инфицировании вирусом гриппа ex-vivo. Потому опосредованное макрофагами, цитокин-индуцированное повреждение легких, может происходить в основном благодаря недавно рекрутированным макрофагам. Тем не менее, множественность ролей цитокинов при инфекции вирусом гриппа и значительная избыточность их сигнальных путей приводят к противоречивым выводам из экспериментального заражения вирусом гриппа мышей, дефицитных по специфическим цитокинам. Остается неясным то, какие индуцированные макрофагами цитокины наиболее важны в повреждении АГБ, является ли это повреждение прямым или непрямым и, собственно, механизм этого повреждения. Таблица 2. Продукция цитокинов после заражения гриппом А моноцитарными клетками и альвеолярными макрофагами человека.*Определяется экспрессией мРНК. TNF = фактор некроза опухоли.
Заключение и перспективы
В этом обзоре мы обозначили основные типы клеток и механизмов, вовлеченных в поражение легких вирусом гриппа, которые ведут к следующей модели индуцированного вирусом гриппа повреждения АГБ (рисунок 4). При входе в альвеолы, вирус гриппа заражает свою основную мишень — эпителиальные клетки, которые претерпевают апоптоз или некроз, нарушая целостность эпителиального слоя. Инфицированные вирусом гриппа эпителиальные клетки также продуцируют цитокины (например CCL2, CCL5, and CXCL10), которые рекрутируют нейтрофилы и моноциты в альвеолы путем хемотаксиса. Активация инфицированных вирусом гриппа эндотелиальных клеток приводит к повышению экспрессии молекул адгезии для экстравазации лейкоцитов и продукции цитокинов, таких как CCL2, CXCL10, интерферон α, интерлейкин 6, TNFα и интерферон γ. Оба процесса дополнительно привлекают макрофаги и нейтрофилы в альвеолы. Рекрутированные нейтрофилы продуцируют активные формы кислорода, которые могут вызвать повреждение тканей. Нейтрофилы также, в ответ на заражение вирусом гриппа, продуцируют цитокины. В частности, продукция CXCL10 может повредить АГБ, спровоцировав выработку супероксида или усилив хемотаксис нейтрофилов. Рекрутированные макрофаги могут повреждать АГБ тремя путями. Во-первых, они могут экспрессировать TRAIL, который взаимодействует с рецептором смерти 5 на эпителиальных клетках и индуцирует апоптоз эпителиальных клеток. Во вторых, за счет активации NOS2, рекрутированные макрофаги могут повышать концентрации оксида азота и пероксинитрита, которые вызывают повреждение тканей. В-третьих, они являются важными производителями провоспалительных цитокинов, которые еще больше усугубляют воспалительный ответ и могут повредить эпителиально-эндотелиальный барьер. Патогенез индуцированного вирусом гриппа ОРДС сосредоточен на альвеолярном эпителии. Этот подход контрастирует с классической моделью патогенеза ОРДС, в которой основной мишенью считался эндотелий сосудов легких. Важной причиной ОРДС в классической модели считался бактериальный сепсис, который отличается от заражения вирусом гриппа по двум важным критериям. Во-первых, типовой патологический агент при сепсисе — липополисахарид — попадает в альвеолы посредством кровотока, в то время как вирус гриппа проникает туда через дыхательные пути. Во-вторых, основной мишенью липополисахарида является клетка эндотелия, в то время как для вируса это клетка эпителия. Неудивительно, что в старых моделях так много внимания было уделено клеткам эндотелия: на многих моделях для изучения патогенеза ОРДС использовали именно липополисахарид. Независимо от того, был ли ОРДС вызван сепсисом, респираторным вирусом или же другим патогеном, его патогенез в основном зависит от повреждения альвеолярного эпителия, который является основным барьером, поддерживающим разность осмотического давления по разные стороны АГБ. Источник: журнал Lancet (А) Схематическая модель патогенеза ОРДС индуцированного вирусом гриппа. В здоровых легких просвет альвеол свободен от жидкости, что обеспечивает оптимальный газообмен (верхний правый квадрант). После инфицирования, вирус гриппа А индуцирует клеточную смерть, что приводит к экссудации белоксодержащей жидкости в просвет альвеол (верхний правый квадрант). Вирус гриппа также индуцирует продукцию цитокинов эпителиальными и эндотелиальными клетками и повышение экспрессии селектина на эндотелиальных клетках. Повышение экспрессии селектина эндотелиальными клетками допускает экстравазацию нейтрофилов, которые являются первыми среди клеток рекрутируемых в легкие при заражении вирусом гриппа (нижний левый квадрант). Нейтрофилы повреждают аэро-гематический барьер (АГБ) посредством токсических гранул и продукции цитокинов, что приводит к увеличению экссудации белоксодержащей жидкости в альвеолярное пространство. Повышение экспрессии селектина на эндотелиальных клетках допускает экстравазацию макрофагов, которые повреждают АГБ посредством продукции NO, TRAIL и цитокинов (верхний левый квадрант). В дополнение к белковой жидкости и лейкоцитарной инфильтрации, прогрессирующее повреждение АГБ приводит к осаждению фибрина и кровоизлияниям в просвете альвеол. (В) Гистопатологический препарат показывает характерные этапы ОРДС в легких хорька через 4 дня после заражения вирусом гриппа А H5N1. Верхний правый квадрант демонстрирует нормальные, заполненные воздухом альвеолы неинфицированного хорька. Нижний правый квадрант показывает альвеолы обильно заполненные экссудатом, визуализируемые как розовый гомогенный материал (стрелка). Нижний левый квадрант демонстрирует альвеолы с экссудатом, который смешан с инфильтрировавшими лейкоцитами, главным образом — нейтрофилами (стрелка). Верхний левый квадрант показывает альвеолы, заполненные экссудатом, смешанным с макрофагами, эритроцитами и нитями фибрина. Кроме того, альвеолярные стенки разрушены (окрашивание гематоксилин-эозином, уменьшено на 24% от x400). ОРДС = острый респираторный дистресс синдром. IAV = вирус гриппа А. NO = оксид азота. TRAIL = фактор некроза опухоли связанный с апоптоз-индуцирующим лигандом. Эта новая модель индуцированного вирусом гриппа ОРДС применима для изучения его патогенеза: как для идентификации тем, представляющих особый интерес, так и для обнаружения “белых пятен” в наших знаниях. Возможно ли воплотить эту модель, данные для которой были получены из различных источников, в единую экспериментальную систему? Путем каких механизмов вирус гриппа индуцирует гибель клеток альвеолярного эпителия? Какие факторы определяют наступление апоптоза или некроза? Как этот механизм зависит от типа клеток (например пневмоцитов 1 или 2 типа)? Путем каких механизмов цитокины, продуцируемые эпителиальными клетками, эндотелиальными клетками и недавно рекрутированными макрофагами, повреждают АГБ? Каковы самые важные механизмы, посредством которых активируются эндотелиальные клетки: прямой контакт с вирусом, продукция цитокинов инфицированными вирусом гриппа эндотелиальными клетками или продукция цитокинов другими типами клеток? На какой стадии повреждения АГБ частицы вируса гриппа выбрасываются в большой круг кровообращения и от каких факторов зависит их выход? Любое ли поражение вирусом гриппа повреждает базальную мембрану? Если да, то как? Какое влияние оказывает это повреждение на реэпителизацию? Какие характеристики индуцированного вирусом гриппа повреждения альвеолярного эпителия определяют, произойдет ли реэпителизация либо фиброз? Как вышеуказанные механизмы различаются среди различных штаммов вируса гриппа? Какие факторы хозяина определяют, разовьется ли у пациента ОРДС? Как разница в основной цели (эпителий или эндотелий), для ОРДС, индуцированного вирусом гриппа, и ОРДС, индуцированного сепсисом, отражается на прогрессировании заболевания, ответе на вмешательство или клиническом исходе? Данные о том, что пациенты с индуцированным сепсисом ОРДС имеют более высокие концентрации прокальцитонина и интерлейкина 6 на 1-2 день после диагностики ОРДС, более тяжелое течение болезни и более высокую смертность, чем пациенты с не-септическим ОРДС говорят в пользу предположения о том, что различные причины ОРДС могут приводить к различным клиническим результатам. Описанная в этом обзоре модель также обеспечивает возможность выбора оптимальной терапевтической мишени для минимизации повреждения тканей в ответ на поражение легочных альвеол вирусом гриппа. Потенциальные терапевтические мишени для индуцированного вирусом гриппа ОРДС — альвеолярный эпителий, инфильтрирующие лейкоциты и общий провоспалительный ответ. Факторы роста, такие как фактор роста кератиноцитов и фактор роста гепатоцитов, помогают защитить и восстановить альвеолярный эпителий. Ингибиторы оси CXCL10–CXCR3 снижают инфильтрацию нейтрофилами и индукцию производства активных форм кислорода. Апокинин, ингибитор НАДФ оксидазы 2, также снижает продукцию активных форм кислорода. Целекоксиб и месалазин, ингибиторы ЦОГ-2, снижают дисфункцию цитокинов и предотвращают апоптоз. Гемифиброзил, агонист пероксисомного активирующего пролиферацию рецептора α, снижает выделение провоспалительных цитокинов. Мезенхимальные стволовые клетки сдерживают воспалительный ответ, улучшают клиренс альвеолярной жидкости и поддерживают целостность легочного эпителия и эндотелия при пневмонии. Стволовые клетки легких быстро распространяются в области повреждения альвеол и экспрессируют характерные для альвеол маркеры, после сублетального заражения мышей вирусом гриппа. Понимание сигналов, которые запускают пролиферацию и распространение этих клеток и наблюдаемую регенерацию альвеолярных структур может быть полезно для разработки новых методов лечения индуцированного вирусом гриппа ОРДС. Кроме того, терапия должна способствовать реэпителизации в отсутствие широко распространенного фиброза (который может привести к снижению функции легких и повышению зависимости от ИВЛ). Тем не менее, процедуры, которые ограничивают индуцированное вирусом гриппа повреждение у мышей, могут не обладать эквивалентными эффектами у людей. Следовательно, в профилактике повреждения альвеол вирусом гриппа необходим подход, который сочетает в себе знания полученные в ходе экспериментального заражения лабораторных животных с клиническими данными. Индуцированный вирусом гриппа ОРДС имеет высокую летальность, несмотря на интенсивную терапию, стратегии вентиляционной защиты легких и использование экстракорпоральной мембранной оксигенации. Терапевтических методик, направленных против вируса зачастую недостаточно для того, чтобы вылечить болезнь, потому что повреждения легких, остающиеся после вирусной инфекции, слишком серьезны, чтобы разрешиться самостоятельно. Только благодаря лучшему пониманию механизмов развития повреждения и репарации могут быть разработаны терапевтические стратегии для улучшения исхода данного трудноизлечимого осложнения гриппа. Перевод: Елена Лисицына Изображения: Елена Лисицына, Антон Осипенко Редакция: Азат Муртазин, Михаил Гусев, Станислав Груздев ОригиналЭкспрессия молекул клеточной адгезии фагоцитами как критерий дифференциальной диагностики гипертензивных расстройств беременных
ФГБУ Ивановский научно-исследовательский институт материнства и детства им. В.Н. Городкова Минздрава России, Иваново
Цель исследования. На основании изучения экспрессии молекул клеточной адгезии нейтрофилами и моноцитами венозной крови у беременных с различными формами артериальной гипертензии (АГ) предложить новые дифференциально-диагностические критерии данной патологии.
Материал и методы. Обследованы 50 женщин с преэклампсией (ПЭ), 36 – с хронической артериальной гипертензией (ХАГ), 33 – с ХАГ с присоединившейся ПЭ и 45 женщин контрольной группы. В крови определялось количество CD11b+, CD49b+, CD99+ моноцитов и нейтрофилов методом проточной цитофлюориметрии.
Результаты. По сравнению с группой контроля при ПЭ выявлено повышение уровня CD49b+ нейтрофилов; при ХАГ – повышение CD99+ нейтрофилов, снижение CD11b+ моноцитов; при ХАГ с ПЭ – повышение CD99+ и CD49b+ нейтрофилов. Исследуемые показатели информативны в дифференциальной диагностике гипертензивных расстройств у беременных.
Заключение: определение уровня CD11b+, CD49b+, CD99+ моноцитов и нейтрофилов можно использовать в качестве дополнительных диагностических критериев при определении формы АГ у беременных.
Артериальная гипертензия (АГ) при беременности – актуальная проблема многих десятилетий и в науке, и в практическом акушерстве. Гипертензивные расстройства осложняют до 30% беременностей [1, 2] и являются неоднородным понятием, включающим в себя несколько нозологических форм – гестационную АГ, преэклампсию (ПЭ), хроническую артериальную гипертензию (ХАГ) и ХАГ с присоединившейся протеинурией (преэклампсией) [3]. Нередко в клинической практике специалистам приходится сталкиваться с трудностями дифференциальной диагностики АГ у беременных. Сложности диагностического поиска могут быть обусловлены несколькими причинами: отсутствием контроля артериального давления женщинами до и во время беременности, физиологической гипотензией на протяжении первого и второго триместра, стабильными значениями артериального давления на фоне гипотензивной терапии у женщин с ХАГ, стертым и малосимптомным течением ПЭ. Таким образом, по данным анамнеза, общему состоянию женщины, уровню артериального давления и протеинурии не всегда удается определить форму АГ, а также оценить истинную тяжесть ПЭ, особенно если она присоединилась к существовавшей ранее гипертензии. Важное медико-социальное значение данной группы осложнений беременности определяет необходимость поиска дополнительных дифференциально-диагностических критериев гипертензивных расстройств у беременных с целью своевременного начала лечения и определения тактики ведения пациенток. Известно, что гипертензивные расстройства у беременных сопровождаются изменениями иммунологических показателей – повышением функциональной активности гранулоцитов, моноцитов, лимфоцитов, активацией системы комплемента, повышением продукции цитокинов [4–6]. Изменение цитокинового профиля с преобладанием провоспалительных фракций (интерлейкин (IL)-6, IL-1β, IL-8, IL-16) сопровождается активацией лейкоцитов и развитием системного воспалительного ответа [7, 8]. Под действием медиаторов воспаления усиливается экспрессия адгезионных молекул на эндотелиоцитах и на циркулирующих клетках крови, что способствует еще большей активации, мобилизации и экстравазации лейкоцитов. Накопление активированных клеток в субэндотелиальном слое и периваскулярном пространстве является основой асептического воспаления и одним из механизмов развития эндотелиальной дисфункции в организме беременной женщины [6, 9]. Вазоспазм и гипоксически-ишемические изменения тканей при эндотелиозе отражают системный характер повреждения сосудистого русла, что клинически проявляется формированием АГ.
Цель исследования: на основании изучения экспрессии молекул клеточной адгезии (CD11b, CD49b и CD99) нейтрофилами и моноцитами венозной крови у беременных с различными формами АГ предложить новые дифференциально-диагностические критерии данной патологии.
Материал и методы исследования
Панова И.А., Кудряшова А.В., Малышкина А.И., Хлипунова Д.А., Рокотянская Е.А.
Экстравазация лейкоцитов — обзор
65.5.6 Движение лейкоцитов через интерстиций
В то время как рекрутирование лейкоцитов изначально опосредовано процессом адгезии лейкоцитов и их трансмиграции через эндотелий, по завершении экстравазации лейкоцитов происходит еще один не менее важный процесс — миграция через интерстициальную ткань. ткани. Этот процесс очень сложен и остается малоизученным; однако одновременно задействованы множественные клеточные ответы, включая дифференциальную регуляцию клеточной адгезии с внеклеточным матриксом, химиочувствительность к направленным химическим и белковым градиентам, регуляцию внутриклеточной организации цитоскелета и модуляцию различных путей передачи сигналов.Они находятся под контролем многочисленных сигналов из местного микроокружения, таких как молекулы адгезии, хемоаттрактанты, белки внеклеточного матрикса и механические силы, интегрированные в клетку для генерации специфических клеточных ответов.
Регуляция миграции лейкоцитов молекулами адгезии . Миграция лейкоцитов происходит за счет сочетания актинового выпячивания и сокращения с динамическим формированием и разборкой адгезий между клетками и внеклеточным матриксом и в значительной степени зависит от адгезии.И образование новых матричных контактов, и генерация тяговых сил опосредуются интегринами. 287–289 Изменение адгезионного состояния клетки вызывает двухфазный эффект на миграцию клеток с низким уровнем адгезии, недостаточным для поддержки тяговых сил, и высоким уровнем адгезии, предотвращающим смещение клеток. Однако изменение формы и размера клеток посредством реорганизации внутриклеточной актиновой сети может также регулировать миграцию лейкоцитов посредством событий, не зависящих от интегрина. 290 В совокупности миграция лейкоцитов через трехмерные матрицы сильно амебоидна, но, в зависимости от состава и архитектуры матрицы, мезенхимоподобная миграция со сложной адгезией и системами нацеливания также используется для обеспечения эффективной направленной миграции к повреждающим или инфекционным источникам. 291
Адгезия интегрина к компонентам внеклеточного матрикса, таким как коллагены, фибронектин и ламинины, в первую очередь ответственна за миграцию нейтрофилов через интерстиций.Сообщалось, что члены семейства интегринов β 1 (члены очень позднего антигена (VLA)) в первую очередь ответственны за адгезию нейтрофилов к матрице без заметной роли интегринов β 2 . 292 293 Интересно, что экспрессия β 1 в циркулирующих популяциях лейкоцитов низкая по сравнению с интегринами β 2 . Однако активация интегрина β 2 посредством связывания с лигандом, сопутствующая экстравазации, может быстро активировать локализацию интегрина β 1 на поверхности нейтрофилов. 293 В соответствии с повышенной экспрессией интегринов β 1 , блокада антител или генетическая делеция β 2 интегрина не изменяет миграцию нейтрофилов через интерстиций или адгезию к матричным белкам, что предполагает наличие дополнительных механизмов для увеличения экспрессии и функции β 1 . 294 295 В совокупности эти сообщения предполагают, что экстравазация нейтрофилов включает превращение β 2 в β 1 , опосредованную интегрином подвижность клеток, которая регулируется взаимодействиями интегрин-лиганд β 2 и последующим вовлечением базальной мембраны эндотелиальных клеток.
Дополнительная регуляция миграции нейтрофилов через интерстиций происходит через дифференциальные α-цепи интегрина. Ассоциация различных гетеродимерных цепей α интегрина (α 1–6 ) с цепью β 1 значительно изменяет подвижность нейтрофилов посредством различных внеклеточных матриц. Например, сообщалось, что α 2 β 1 играет главную роль в хемотаксисе нейтрофилов через внесосудистую ткань, такую как брыжейка, в основном за счет взаимодействия с коллагеном. 296 Интересно, что миграция нейтрофилов через гели коллагена с добавлением фибронектина или тенасцина существенно снижена по сравнению с одним коллагеном. 297 298 Более тщательное изучение этого ответа показало, что ингибирование антителами α 4 β 1 и α 5 β 1 интегринов восстанавливает подвижность нейтрофилов посредством комбинированных матриц, как только коллаген, что позволяет предположить, что дифференциальное взаимодействие интегринов с различными матрицами компоненты регулируют общее адгезионное состояние нейтрофилов. 298 Вместе эти исследования предполагают важную роль регулируемого интегрином гемотаксиса, который может участвовать в контроле локализации нейтрофилов в тканях.
Миграция Т-клеток через интерстиций может использовать интегрины β 1 и другие неизвестные механизмы. 288,292,299 Однако также существуют уникальные молекулярные механизмы, регулирующие подвижность Т-клеток. 287–289 Вовлечение CD3 или CD3 / CD2 в Т-клетки может значительно усиливать β 1 интегрин-зависимую адгезию матрикса за счет активации множественных сигнальных путей, которые могут изменять сродство / авидность интегрина и цитоскелетные взаимодействия. 299–301 В частности, стимуляция CD3-TCR приводит к активации киназы Lck / Zap-70, которая увеличивает активность киназы PI3, которая усиливает локализацию тирозинкиназы Itk на мембране, где она может фосфорилироваться с помощью Lck для изменения полимеризации актина, таким образом модулируя β 1 интегриновая функция. Миграция Т-клеток также уникальна тем, что β 2 интегрин LFA-1 (α L β 2 ) участвует в усилении подвижности Т-клеток. LFA-1 повсеместно экспрессируется на всех Т-клетках и играет важную роль в хоминге, рекрутинге и формировании иммунологических синапсов. 302,303 Однако исследования показали, что активация LFA-1 антителами или лигандом (например, ICAM-1) усиливает подвижность Т-клеток на некоторых белках внеклеточного матрикса, включая фибронектин, ламинин и коллаген IV. 304,305 LFA-1 усиленная подвижность Т-клеток также включает активацию внутриклеточных сигналов, включая кальмодулин / MLCK, PKC-β1, рецепторы, связанные с G-белком, и пути RhoA / ROCK. 221,223 Эти отчеты демонстрируют, что подвижность Т-клеток очень динамична и происходит таким образом, чтобы эффективно исследовать ткань на наличие антигена.
Т-клетки памяти, выделенные из тканевого микроокружения, а не из периферической крови, также обнаруживают дифференциальную экспрессию интегрина, которая изменяет адгезию между клетками и внеклеточным матриксом. В исследовании Krivacic и Levine изучались профили экспрессии интегрина и адгезия к природному внеклеточному матриксу или его компонентам между Т-клетками собственной пластинки или Т-клетками периферической крови, которые культивировались и активировались in vitro. 306 В этом исследовании было сообщено о нескольких важных выводах. Во-первых, периферические Т-клетки показали меньшую адгезию к внеклеточному матриксу по сравнению с Т-клетками собственной пластинки.Во-вторых, периферические лимфоциты проявляли преимущественное связывание с фибронектином, в то время как лимфоциты собственной пластинки предпочли коллаген. В-третьих, дифференциальное связывание матрикса было связано с большим процентом Т-клеток собственной пластинки, экспрессирующих VLA α интегриновые цепи 1, 2, 3 и 5. Это не было связано с различиями в экспрессии субъединиц на поверхности на индивидуальной клеточной основе. Наконец, длительная экспансия IL-2 в лимфоцитах периферической крови, культивируемых на матриксных белках, вырабатывала экспрессию поверхностного интегрина и характеристики адгезии, аналогичные характеристикам лимфоцитов собственной пластинки.Эти находки предполагают, что окружающая среда местного тканевого матрикса может дополнительно дифференцировать рекрутированные Т-лимфоциты по фенотипу с высокой адгезией, способствующим длительной секвестрации в ткани.
Миграция моноцитов в интерстиции также включает α 1 β 1 (VLA-1). Исследования с использованием блокады антител или целенаправленного генного удаления цепи интегрина α 1 демонстрируют значительную защиту от декстрансульфата натрия или колита, индуцированного TNBS. 307,308 Интересно, что ингибирование функции α 1 преимущественно подавляло инфильтрацию моноцитов в интерстиций, что не было связано со снижением рекрутирования иммунных клеток, как определено с помощью прижизненной микроскопии. 308 Эти отчеты демонстрируют, что экспрессия α 1 β 1 важна для адгезии моноцитов к внеклеточному матриксу и последующего движения через интерстиций. В совокупности все вышеупомянутые исследования показывают, что однократное или множественное ингибирование ассоциации белков VLA с внеклеточным матриксом представляет собой уникальный терапевтический подход к воспалительным расстройствам.
Хемокиновая регуляция движения лейкоцитов через интерстиций : Процесс химиочувствительности лейкоцитов во время воспалительной реакции важен для обеспечения оптимального рекрутирования определенных типов лейкоцитов в отдельные участки ткани и органы. Было описано множество молекул, которые способствуют направленной миграции лейкоцитов, включая хемокины, цитокины и другие биохимические медиаторы (например, fMLP, PAF, продукты комплемента, метаболиты арахидоновой кислоты и другие).
Исследования последних нескольких лет позволили получить важное физиологическое и патологическое понимание критической роли хемокинов в регулировании рекрутирования лейкоцитов из сосудистой сети и их последующей миграции через ткань. Некоторые хемокины участвуют в нарушении регуляции иммунных ответов желудочно-кишечного тракта. Сообщалось о повышении экспрессии хемокинов CXCL5, CXCL8, CXCL10, CCL2, CCL5, CCL6 и CCL10 у пациентов с активным язвенным колитом (UC) или болезнью Крона (CD). 309,310 Аналогичным образом, повышенная экспрессия хемокинового рецептора CXCR1 для CXCL8 и CXCR3 для CXCL10 также наблюдается в ткани толстой кишки пациентов с ВЗК. Важная патологическая роль хемокинов была дополнительно продемонстрирована на животных моделях экспериментального колита. Повышенная экспрессия хемокинов CXCL1, CCL2, CCL3 и CCL5 была обнаружена в различных моделях колита. 310 В соответствии с этими наблюдениями, генная делеция CCR2, CCR5 или CCR6 ослабляет экспериментальный колит, предполагая, что усиленная экспрессия и высвобождение хемокинов вызывает рекрутирование лейкоцитов, несущих специфические хемокиновые рецепторы. 311,312 Вместе эти данные демонстрируют важную патологическую роль хемокинов и их рецепторов во время воспаления ЖКТ.
Хемокины могут способствовать привлечению и миграции лейкоцитов за счет изменения функции адгезионных молекул или клеточного местоположения. Например, сообщалось, что несколько хемокинов, включая CXCL8, CXCL9, CXCL10, CXCL12, CCL2, CCL11, CCL17, CCL19, CCL21 и CCL22, стимулируют быструю опосредованную интегрином твердую адгезию лейкоцитов в условиях гидродинамического потока. 313 Стимуляция хемокинами усиливает адгезию, опосредованную интегрином, путем изменения состояния активации интегрина (переход от «закрытой» несвязывающей конформации к «открытому» состоянию с высоким уровнем связывания), тем самым увеличивая его сродство к лиганду. Они могут дополнительно изменять расположение на поверхности интегриновых клеток в дискретных кластерах, тем самым увеличивая авидность лиганда. 314,315 Эти молекулярные события могут также происходить одновременно в лейкоцитах, что объясняет очень быструю и эффективную природу хемокин-опосредованной адгезии лейкоцитов. 316 Хемокиновая активация лейкоцитов также увеличивает адгезию к белкам внеклеточного матрикса посредством аналогичных механизмов и стимулирует устойчивую миграцию через внеклеточный матрикс. 298,317–321 Однако хемокины — не единственный компонент воспалительной среды хемоаттрактанта, и остается неясным, как эти молекулы действуют совместно с другими цитокинами, стимулируя тканевую инфильтрацию лейкоцитов.
Подобно хемокинам, цитокины могут также связываться с матриксными белками и создавать «хемоаттрактантный след» для лейкоцитов. 322 Например, TNF-α связывается с N-концевым доменом фибронектина, который усиливает связывание Т-клеток с фибронектином в зависимости от интегрина β 1 . 323 Точно так же TGF-β также модулирует адгезию лейкоцитов и миграцию через матричные белки. 324,325 В предварительных исследованиях сообщалось о том, как комбинаторные градиенты цитокинов и хемокинов влияют на миграцию лейкоцитов через внеклеточный матрикс. Эксперименты с использованием трехмерных внеклеточных матриц, содержащих цитокин (например,g., TNF-α или IL-2) к градиентам хемокина (например, CXCL12 или CCL5) показывают, что цитокиновая стимуляция ингибирует хемотаксис лимфоцитов из-за повышенной адгезии интегрина к матриксным белкам. 326 Важно отметить, что адгезионный ответ, стимулированный комбинированной стимуляцией цитокином / хемокином, существенно выше, чем адгезия, опосредованная только цитокином, что предполагает синергетический эффект этих хемоаттрактантов в усилении взаимодействия клетки с матрицей. Интересно, что эффекты комбинированного лечения хемокинами могут фактически противодействовать клеточным ответам друг на друга, включая снижение хемотаксических ответов и адгезию к матрице. 327 Эти наблюдения показывают, что биофизическая регуляция миграции лейкоцитов через ткань является многофакторной и предполагает, что дифференциальное рекрутирование лейкоцитов во время различных воспалительных реакций включает несколько членов семейства цитокинов и хемокинов.
Трансэндотелиальная миграция нейтрофилов: обновления и новые перспективы | Кровь
Параклеточный диапедез сам по себе является многоступенчатым процессом, который включает в себя участие нескольких молекул адгезии, в том числе ICAM-1/2, молекулы адгезии сосудистых клеток 1, молекулы соединительной адгезии A (JAM-A) и JAM-C, PECAM-1, CD99, и молекула адгезии, избирательная к эндотелиальным клеткам. 5,56 Это было продемонстрировано исследованиями с использованием генетических моделей мышей и прижизненной микроскопии, которые определили точное место, где трансмиграция лейкоцитов блокируется. После прочной адгезии взаимодействия между нейтрофилами и ЭК происходят через ICAM-1 и β2-интегрин, что, в свою очередь, запускает ослабление прикрепленных контактов ЭК за счет фосфорилирования сосудисто-эндотелиального кадгерина. 57 Впоследствии лейкоциты мигрируют через соединение EC посредством последовательного взаимодействия с JAM-A 58,59 и затем с PECAM. 60 Точная роль PECAM при диапедезе остается спорной. Например, PECAM-дефицитные нейтрофилы мыши в основном задерживались на уровне периваскулярной базальной мембраны in vivo. 58,59 Однако исследования нейтрофилов человека показали, что нейтрализующие антитела PECAM задерживают нейтрофилы между ЭК in vitro. 61,62 Другие важные эксперименты с использованием последовательного добавления и удаления анти-PECAM и анти-CD99 блокирующих антител или наоборот предполагают, что CD99 требуется на более поздней стадии процесса трансмиграции, чем PECAM. 61,62 Блокирование CD99 или CD99L2 нейтрофилов in vivo между ЭК и нижней базальной мембраной в воспаленной ткани кремастера. 63 Недавно группа Мюллера пролила свет на эти кажущиеся несоответствия. Они использовали 4-мерную прижизненную микроскопию, чтобы показать, что место и порядок функционирования PECAM и CD99 in vivo зависят от линии мышей. У мышей FVB / n PECAM функционирует выше CD99, как в клетках человека in vitro, и блокирует антитела против любой из молекул, задерживая нейтрофилы до того, как они пройдут через эндотелий. 64 Однако у мышей C57BL / 6 те же антитела останавливают миграцию лейкоцитов через базальную мембрану эндотелия. 64 Интересно, что взаимодействия PECAM-1 стимулируют рекрутирование нелигированных молекул адгезии (например, PECAM, JAM-A и CD99), с которыми лейкоциты могут взаимодействовать внутри эндотелиального соединения, вероятно, направляя лейкоциты, перемещающиеся через соединение. Нелигированные молекулы привлекаются к границе ЭК через определенные типы везикул, которые называются компартментом рециклинга боковой границы эндотелия (LBRC). 65 Наконец, во время этого процесса нейтрофилы генерируют ядерные доли, которые вставляются в ламеллиподии в передней части клеток, которые изгибают эндотелиальную актиновую сеть и проталкиваются через барьер ЭК. 66 Сократительные стрессы, вызываемые нейтрофилами и ЭК, также необходимы для нарушения напряжений эндотелиальных соединений, чтобы открыть промежутки для трансмиграции и для нейтрофилов, чтобы протолкнуть себя через промежуток. 67 Следовательно, диапедез может регулироваться механически путем трансмиграции лейкоцитов и провоспалительных сигналов, которые увеличивают сократимость ЭК.
Недавнее исследование проливает свет на плохо определенную роль хемокиновых рецепторов. Группа Nourshargh только что сообщила, что CXCL1, который продуцируется в основном стимулированными фактором некроза опухолей ECs и перицитами, поддерживает ползание нейтрофилов в просвете и суб-EC. Напротив, нейтрофилы являются основными продуцентами CXCL2, и этот хемокин имеет решающее значение для правильного нарушения соединений EC. 68
общих и уникальных механизмов, используемых различными подгруппами лейкоцитов во время экстравазации
Экстравазация лейкоцитов — один из основных и первых шагов во время инициации воспаления.Следовательно, лучшее понимание ключевых молекул, регулирующих этот процесс, может помочь в разработке новых терапевтических средств для лечения заболеваний, связанных с воспалением, таких как атеросклероз или ревматоидный артрит. Молекулы эндотелиальной адгезии ICAM-1 и VCAM-1 известны как центральные медиаторы адгезии лейкоцитов к эндотелию и их миграции через него. Вовлечение этих молекул их рецепторами интегрина лейкоцитов инициирует активацию нескольких сигнальных путей как в лейкоцитах, так и в эндотелии.Было описано, что некоторые из таких событий происходят во время трансэндотелиальной миграции всех субпопуляций лейкоцитов, тогда как другие механизмы известны только для одной субпопуляции лейкоцитов. Здесь мы обобщаем современные знания о регуляторных механизмах экстравазации лейкоцитов с лейкоцитарной и эндотелиальной точки зрения соответственно. В частности, мы сосредоточимся на выявлении общих и уникальных механизмов, которые используют определенные субпопуляции лейкоцитов для успешного пересечения эндотелиальных монослоев.
1.Введение
Воспалительная реакция имеет решающее значение для борьбы с инфекциями и заживления ран и, таким образом, необходима для выживания [1, 2]. Однако постоянно активные иммунные реакции предшествуют хроническим воспалительным расстройствам и другим патологиям. Таким образом, необходимо строго контролировать иммунный ответ на травму и инфекцию. Чтобы специфически препятствовать чрезмерной трансэндотелиальной миграции лейкоцитов (ТЕМ), требуется детальное понимание регуляции этого многоступенчатого процесса.Бутчер и Спрингер предложили во вневременных обзорах многоступенчатую модель процесса ПЭМ [3, 4]. В настоящее время эта предложенная модель все еще действует; однако со временем к последовательности событий во время ТЕА были добавлены некоторые дополнительные шаги [2]. Воспалительная реакция начинается с секреции провоспалительных медиаторов, таких как гистамин или цитокины, которые вызывают открытие контактов эндотелиальных клеток (ЭК) в посткапиллярных венулах, чтобы обеспечить прохождение молекул крови, например факторов комплемента.Воспаление также включает поверхностную экспрессию молекул адгезии эндотелия, ремоделирование актина и активацию интегринов лейкоцитов, которые обеспечивают адгезию лейкоцитов на эндотелий внутри сосудистой стенки и последующий диапедез [5-8]. Последовательность адгезионных взаимодействий лейкоцитов с ЭК называется каскадом экстравазации лейкоцитов и включает серию адгезивных взаимодействий, которые позволяют сначала привязать, перекатывать и медленно перекатывать, за которыми следует прочная адгезия, ползание и образование трансмиграционной чашечки на апикальной эндотелиальной поверхности (рис. 1).Далее идет фактическая ТЕА лейкоцитов (также называемая диапедезом), которая может происходить при пересечении контактов ЭК (параклеточная) или тела ЭК (трансцеллюлярная). Оба пути существуют, и известно, что сила эндотелиальных соединений контролирует предпочтение маршрута [9], но точные лежащие в основе механизмы остаются неуловимыми. После прохождения через эндотелий лейкоциты также должны пересечь слой перицитов и базальную мембрану (BM), чтобы достичь воспаленной ткани и способствовать избавлению от инфекции и заживлению ран [10].Различные типы лейкоцитов привлекаются к участкам воспаления, включая нейтрофилы, моноциты и лимфоциты. В ответ на воспалительный стимул нейтрофилы обычно находятся среди первых лейкоцитов, покидающих кровоток, и после дегрануляции они вносят вклад во вторую волну трансмиграции, главным образом моноцитами [11]. Также наблюдался обратный случай, когда присутствие моноцитов и хемоаттрактантов нейтрофилов, происходящих из моноцитов, требовалось для рекрутирования нейтрофилов в места стерильного воспаления [12].Рекрутирование всех этих субпопуляций лейкоцитов является обязательным для правильного иммунного ответа, поскольку все они выполняют разные функции, однажды задействованные в воспаленной ткани [13]. Все эти типы лейкоцитов следуют последовательным этапам каскада экстравазации в целом, но были описаны различия в реакции на определенные хемокины и в экспрессии / активации молекул адгезии, которые опосредуют взаимодействия с EC [8, 14]. Было подтверждено, что некоторые механизмы во время каскада экстравазации лейкоцитов, такие как определенные взаимодействия рецептор-лиганд или сигнальные пути, используются всеми субпопуляциями лейкоцитов.Однако другие механизмы пока описаны только для одного типа лейкоцитов. Являются ли эти механизмы действительно уникальными для данной субпопуляции лейкоцитов или они просто еще не были изучены в других субпопуляциях лейкоцитов — важный вопрос, на который предстоит ответить в будущем. Было опубликовано множество обзоров, в которых суммированы некоторые аспекты рекрутирования лейкоцитов, но в обобщенной форме, в которой говорится только о «лейкоцитах». В этом обзоре мы обобщаем текущие знания об общих и уникальных механизмах, которые различные типы лейкоцитов, такие как нейтрофилы, моноциты и лимфоциты, используют во время экстравазации (таблица 1).Это включает в себя сигналы, индуцированные внутри каждой субпопуляции лейкоцитов, а также дифференциальные сигналы, которые каждая субпопуляция лейкоцитов индуцирует в ЭК для облегчения трансмиграции.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Представляет ~ 40–60% лейкоцитов, циркулирующих в крови человека, со скоростью ~ 1 -2 × 10 11 клеток в день в кровоток и с продолжительностью жизни всего 1–5 дней [15], нейтрофилы являются одними из первых лейкоцитов, которые попадают в места воспаления и / или повреждения.Миграция этих уникальных лейкоцитов через стенки кровеносных сосудов — это строго регулируемый процесс, для которого некоторые из молекулярных взаимодействий с различными компонентами стенки сосуда (например, эндотелием, оболочкой перицитов и венулярным костным мозгом) относительно хорошо описаны в литературе. [5, 14, 16]. В настоящее время рассматриваются 5 основных этапов рекрутирования нейтрофилов, а именно: (1) захват и катание вдоль просветной стороны эндотелия, (2) прочная адгезия и ползание к месту проведения ПЭМ, (3) ПЭМ (и его вариации). ), (4) субэндотелиальное ползание вдоль отростков перицитов и (5) выход во внесосудистое пространство через промежутки перицитов и специфические области внутри сосудистой костной ткани.В течение многих десятилетий предполагалось, что хемокины и другие растворимые хемоаттрактанты ответственны за специфичность рекрутирования субпопуляций лейкоцитов из-за уникального репертуара рецепторов, связанных с G-белком, присутствующих на их поверхности [17-19]. Однако недавние убедительные доказательства in vivo поставили под сомнение эту идею и продемонстрировали роль многих адгезионных молекул, присутствующих на поверхности EC, специфически инструктирующих нейтрофилы экстравазировать [20–22].
2.1. Capture and Rolling
Свободно текущие нейтрофилы изолированы от эндотелия плотным слоем 0.От 5 до 5 мкм толщиной м, сеть отрицательно заряженных протеогликанов, гликозаминогликанов и гликопротеинов, называемая гликокаликсом ЭК [23]. Эта структура действует как грозный барьер для эмиграции лейкоцитов и превышает размеры молекул клеточной адгезии, участвующих в рекрутировании нейтрофилов. Следовательно, изменения гликокаликса ЭК являются предпосылкой для первых шагов экстравазации нейтрофилов [24, 25]. Экспрессия положительно заряженных молекул, таких как МПО, на поверхности нейтрофилов [26], а также отщепление гликокаликса ЭК гепариназой [27], высвобождение реактивных форм кислорода, полученных из нейтрофилов (ROS) [28], и матричных металлопротеиназ (MMP). ) [29] способствуют облегчению контактов нейтрофилов с ЭК.После удаления гликокаликса ЭК нейтрофилы могут достигать эндотелиальной поверхности через определенный класс из 3 тесно связанных гликопротеинов, называемых селектинами, и их гликоконъюгатные лиганды (лиганд-1 гликопротеина P-селектина (PSGL-1), CD44 и лиганд E-селектина -1 (ESL-1)) [21, 30, 31]. L-селектин конститутивно экспрессируется на поверхности нейтрофилов, тогда как P- и E-селектины более специфичны для EC. Р-селектин постоянно хранится в отдельных гранулах ЭК, называемых тельцами Вейбеля-Паладе, которые быстро мобилизуются на поверхность ЭК, где Р-селектин равномерно распределяется по клеткам.Напротив, E-селектин синтезируется de novo во время активации и концентрируется в основном в соединениях EC. Интересно, что новое исследование Zuchtriegel et al. [22] продемонстрировало, что нейтрофилы в основном используют P- / L-селектин и PSGL-1 / CD44, но не E-селектин, чтобы связывать и катиться вдоль эндотелия in vivo . При блокировании эти взаимодействия влияли не только на поток катящихся нейтрофилов, но и на их последующую прочную адгезию, ползание и ТЕМ. Напротив, воспалительные моноциты дополнительно должны взаимодействовать с E-селектином для правильной трансмиграции через стенку сосуда, что подчеркивает новую особую разницу в молекулярных взаимодействиях между различными подгруппами лейкоцитов и ЭК во время этой первой стадии трансмиграции.
Несмотря на слабый и временный характер молекулярных взаимодействий между селектинами и их лигандами, нейтрофилы катятся даже при высоком напряжении сдвига внутри кровеносных сосудов. Сандд и его коллеги недавно сделали несколько интересных наблюдений о том, как эти нейтрофилы поддерживают контакт с ЭК во время перекатывания [32]. Во время начальных контактов с EC через селектины / лиганды селектина структура клеточной мембраны нейтрофилов модифицируется путем реорганизации как цитоскелета, так и молекул поверхностной адгезии, что приводит к образованию протяженного выступа, называемого слингом [33].Эта структура образуется из мембранного троса на задней стороне катящегося нейтрофила как якорь, прежде чем она обернется вокруг катящегося лейкоцита и повернется к передней части клетки, чтобы восстановить контакт с ЭК. Такие слинги содержат гетерогенные участки PSGL-1, придающие прерывистые адгезивные структуры поверхности ЭК, но также богаты антигеном-1, ассоциированным с функцией β 2-интегрина лимфоцитов (LFA-1). Более того, связывание PSGL-1 с P- / L-селектином во время стадии сворачивания приводит к конформационным изменениям в нейтрофилах β 2-интегрина LFA-1 посредством передачи сигналов извне-внутрь [34-36].Этот ответ делает возможным связывание LFA-1 с его лигандами на ЭК, поддерживая медленное вращение и, в конечном итоге, переход к прочной адгезии нейтрофила [37].
2.2. Firm Adhesion and Crawling
Усиление взаимодействий нейтрофилов с EC происходит в основном за счет связывания лейкоцитарных 2-интегринов LFA-1 и макрофагального антигена-1 (Mac1) с их родственной рецепторной молекулой межклеточной адгезии-1 (ICAM-1), экспрессируемой на активированном ЭК [38]. Эти взаимодействия позволяют нейтрофилам прочно прикрепляться к поверхности эндотелия.Параллельно с адгезивными комплексами 2-интегрины / ICAM-1, нейтрофильный 1-интегрин очень поздний антиген-4 (VLA-4) и его партнер по связыванию EC молекула адгезии сосудистых клеток-1 (VCAM-1) могут способствовать остановке лейкоциты при определенных воспалительных состояниях человека [39]. Эта усиленная адгезия завершается восприятием хемотаксических молекул, таких как хемокины (например, CXCL1 / 2), липидных медиаторов (например, LTB4, PAF) и белков комплемента (например, C5a) рецепторами, связанными с G-белками (GPCR). на поверхности нейтрофилов, которые далее сигнализируют через цитоскелет, чтобы вызвать полную активацию интегринов и прочную адгезию [37].После этой прочной адгезии нейтрофилы ползут перпендикулярно потоку крови или даже против него, в направлении хемотаксических [40] (например, хемокинов) или гаптотаксических (например, ICAM-2) градиентов. Механизм этого ползания просвета строго зависит от ICAM-1 / Mac1 [41, 42], поскольку блокада этих двух молекул in vivo приводит к тому, что нейтрофилы не могут ни ползать, ни мигрировать через соединения EC, не затрагивая адгезию нейтрофилов. Было высказано предположение, что переход между LFA-1-зависимой прочной адгезией и Mac1-зависимым ползанием нейтрофилов происходит посредством передачи сигналов наизнанку через LFA-1 и активации фактора обмена гуанина Vav-1 [43], который, следовательно, активирует Mac1. [44].Недавно было показано, что другой член семейства CAM, ICAM-2, играет роль в динамике ползания нейтрофилов к соединениям EC до TEM [45]. У мышей с генетической делецией этой молекулы, а также у животных WT, получавших блокирующее антитело против ICAM-2, нейтрофилы демонстрировали увеличение продолжительности ползания и снижение скорости ползания, что приводило к тому, что нейтрофилы дольше задерживались на просветной поверхности ЭК и задерживались. миграция через эндотелиальные соединения.
2.3. ТЕА и его вариации
ТЕА — это самый быстрый ответ каскада миграции нейтрофилов, продолжающийся 5–10 минут в зависимости от воспалительного сценария. Несколько молекулярных взаимодействий между нейтрофилами и ЭК были описаны для этой стадии в литературе [5, 14, 16]. Проникновение EC нейтрофилами происходит двумя путями: через межклеточные соединения EC-EC (т.е. параклеточная миграция) или через тело EC (т.е. трансцеллюлярная миграция). Недавние доказательства in vivo показали преобладание параклеточного пути (90% событий трансмиграции) над трансцеллюлярной миграцией [46].Генетически модифицированные мыши, у которых стабилизированы адгезивные соединения и, в частности, комплекс VE-кадгерин-катенин / VE-PTP, показали, что стенка кровеносных сосудов стала непроницаемой для макромолекул и инфильтрации нейтрофилов [47, 48]. Напротив, мыши, дефицитные по актин-связывающему белку cortactin, демонстрировали снижение кластеризации ICAM-1 вокруг адгезивных нейтрофилов из-за дефектной активации GTPase RhoG в EC, что приводило к сильному снижению адгезии и трансмиграции [49, 50]. Многочисленные молекулы адгезии, обогащенные в соединениях EC-EC, такие как PECAM-1, члены семейства JAM, ICAM-2, CD99, ESAM и CD99L2, участвуют в процессе ТЕА нейтрофилов.Эти молекулы также обнаруживаются в субклеточных структурах, называемых компартментом рециклирования боковой границы (LBRC), которые играют ключевую роль в ТЕА нейтрофилов [51, 52]. В базовых условиях эти молекулы адгезии способствуют поддержанию соединений EC; однако во время воспаления они взаимодействуют со своими контррецепторами на нейтрофилах (например, 2-интегринами LFA1 и Mac1 и посредством гомофильных взаимодействий PECAM-1, JAM-A или CD99, которые также экспрессируются на лейкоцитах), чтобы обеспечить пересечение переходы последовательно [16, 53, 54].Связывание молекул адгезии между нейтрофилами и ЭК может также опосредовать сигналы поляризации в нейтрофилах, позволяя им правильно мигрировать от просветной к аблюминальной сторонам ЭК. Это особенно верно для JAM-A [55] и JAM-C [56]. Две недавние публикации продемонстрировали in vivo наличие аномальных трансэндотелиальных миграционных событий [46, 57], характеризующихся частичной миграцией нейтрофилов через соединение с колебательными движениями в соединении (т.е.е., нерешительная миграция) или даже возвращение в кровоток в направлении от просвета к просвету (обратная миграция) после повреждения ишемией-реперфузией или воспаления, вызванного лейкотриеном B4- (LTB4-). Эта аномальная трансмиграция может составлять до 20% от общего числа событий ТЕА. Этот ответ может быть воспроизведен или даже усилен при других воспалительных состояниях в отсутствие или путем блокады JAM-C [46]. Аномальная трансмиграция действительно включает удаление JAM-C из соединения посредством расщепления нейтрофильной эластазой [57] после его транслокации из азурофильных гранул на поверхность лейкоцита в комплексе с интегрином Mac1 при прямой стимуляции нейтрофила LTB4 [58]. ].Генетическая делеция или фармакологическое ингибирование NE может значительно восстановить присутствие JAM-C на стыках и уменьшить количество аномальных трансмиграционных событий. С другой стороны, экзогенная инъекция NE в воспалительных моделях, которые, как известно, не демонстрируют аномальных ТЕМ нейтрофилов, увеличивала количество этих событий. Этот специфический аномальный ответ ТЕА был связан с присутствием растворимого JAM-C в сыворотке и увеличением вторичного повреждения органов, двумя ключевыми особенностями, которые регулярно наблюдаются у пациентов с травмой или ишемическим реперфузионным повреждением.
2.4. Abluminal Crawling
Более ранние наблюдения за миграционными процессами показали, что после ТЕА стенка сосуда утолщалась и нейтрофилы могли быть обнаружены в ткани только спустя более 20-40 минут после того, как ТЕА произошла. В течение многих десятилетий ничего не было известно о том, что происходило с нейтрофилами в этот период времени. После миграции через ЭК аблюминальный нейтрофил сталкивается со вторым клеточным компонентом, то есть с перицитами, и его плотным матриксом, венулярной базальной мембраной (БМ), в которую они встроены [59].Во многих исследованиях событий трансмиграции эти два компонента стенок кровеносных сосудов не учитывались из-за трудности воспроизведения полной структуры in vitro или ее визуализации in vivo . Однако недавние разработки новых передовых методов микроскопии и поколение генетически флуоресцентных животных пролили новый свет на роль перицитов в рекрутинге нейтрофилов in vivo . Перициты экспрессируют молекулы адгезии и хемокины, такие как ICAM-1, VCAM-1 и CXCL1, при воспалении как in vivo, , так и in vitro [60–63].Этот ответ коррелировал с наблюдениями, что после ТЕА нейтрофилы, как было обнаружено, ползли вдоль перицитов от своего места ТЕА в зависимости от ICAM-1 / Mac1- (и в меньшей степени LFA-1), прежде чем полностью прорваться через венулярный канал. стена [63]. Блокирование этих молекулярных взаимодействий с помощью блокирующих антител может подавлять как аблюминальную подвижность нейтрофилов, так и их проникновение в интерстициальное пространство.
2,5. Выход из стенки сосуда
После ползания по просвету нейтрофилы выходят из стенки сосуда через определенные увеличенные промежутки между соседними перицитами.Роль увеличения промежутков в периците до сих пор неясна, но, что интересно, менее 10% промежутков использовались мигрирующими нейтрофилами, и большую часть времени можно было наблюдать горячие точки трансмиграции, когда более 2-3 нейтрофилов выходили через один и тот же перицит. пробелы. Было высказано предположение, что потенциальное обогащение молекулами адгезии и хемокинами вокруг определенных промежутков перицита [63], а также высвобождение / генерация хемоаттрактантов ведущими нейтрофилами из их гранул [11] и / или от расщепления белков BM на хемотаксические фрагменты [64] могут проложить путь для последующих нейтрофилов.
Венулярный BM (генерируемый как EC, так и перицитами) является конечной интерактивной матрицей (но также и барьером) для эмиграции нейтрофилов. Эта структура состоит из плотных сетей матричных белков, таких как коллаген IV типа и ламинины [65]. Интересно, что блокирование взаимодействий между интегринами лейкоцитов VLA-3 и VLA-6 (рецепторы коллагена и ламинина, соответственно) и венулярным BM с помощью блокирующих антител может ингибировать миграцию нейтрофилов через этот слой [66–68].Другой уникальной характеристикой взаимодействия нейтрофилов с венулярным костным мозгом является обнаружение участков низкой экспрессии (LER) внутри костного мозга, которые являются предпочтительными сайтами миграции нейтрофилов [10, 59]. Эти сайты содержат небольшое количество матричных белков, связаны с промежутками между соседними перицитами и используются и увеличиваются во время миграции нейтрофилов, но не моноцитов [69]. Фактически, нейтрофилам потребуется еще 10-20 минут, чтобы мигрировать через промежутки перицита и LER, как это наблюдалось in vivo , со многими колебательными движениями нейтрофилов [63].Однако продолжительность проникновения LER / перицита в промежуток и колебательных движений для последующих нейтрофилов, следующих за той же горячей точкой миграции, сокращается. Хотя механизм ремоделирования таких пермиссивных сайтов в костном мозге венулярных стенок до конца не изучен, было высказано предположение, что может быть задействовано протеолитическое расщепление нейтрофильными ферментами [10, 59] и / или обратимая разборка коллагеновых волокон [65]. в этом процессе, позволяя нейтрофилам наконец получить доступ к интерстициальному пространству.
3. Механизмы, используемые моноцитами для достижения ТЕА
3.1. Популяции моноцитов
Моноциты — это гетерогенные клетки, которые циркулируют в крови в различных популяциях, называемых резидентными (или патрулирующими) и воспалительными моноцитами в соответствии с профилем экспрессии определенных хемокиновых рецепторов и молекул адгезии [70–72]. В то время как резидентные моноциты связаны с иммунным надзором и заживлением ран, воспалительные моноциты связаны с индукцией и поддержанием воспалительных иммунных ответов [73].С другой стороны, моноциты дают начало дендритным клеткам и макрофагам, способствующим воспалительным ответам [74, 75]. Моноциты активно рекрутируются из костного мозга через кровоток в воспаленные ткани в значительной степени зависимым от CC-хемокинов рецептором 2 / CC-лиганд-2- (CCR2 / CCL2) способом. Совсем недавно элегантное исследование прижизненной визуализации сообщило о фенотипической конверсии субпопуляций моноцитов в участках стерильного повреждения печени [76]. Во-первых, воспалительные моноциты быстро набирались и оставались вокруг поврежденного участка в течение примерно 48 часов, прежде чем произошло преобразование в фенотип резидентных моноцитов и проникновение в поврежденный участок, чтобы вызвать заживление раны.Эта ранее неизвестная пластичность моноцитов подчеркивает важность моноцитов для разрешения воспалений. Кроме того, на мышах был описан подход целенаправленного подавления с использованием наночастиц, содержащих CCR2-специфическую siRNA, которые предотвращали накопление воспалительных моноцитов в местах воспаления и улучшали различные патологические состояния, в которые были вовлечены воспалительные моноциты [77]. Это многообещающий подход для целенаправленного воздействия на воспалительные моноциты без воздействия на другие иммунные клетки во время воспаления; однако еще предстоит доказать, применим ли такой подход к людям.
3.2. Rolling and Slow Rolling
ТЕМ моноцитов происходит в соответствии с парадигмой каскада экстравазации лейкоцитов, как описано выше [78]. Помимо четко установленной роли PSGL-1 во всех свертываниях лейкоцитов, прокатка моноцитов во время рекрутирования в лимфоидные ткани также зависит от L-селектина и CD44 [79]. В инфицированной коже правильное сворачивание моноцитов и последующее рекрутирование зависели от взаимодействия моноцитов PSGL-1 с эндотелиальным E- и P-селектином, тогда как L-селектин моноцитов взаимодействовал с адрессином эндотелиальных периферических узлов (PNAd) [80].Шеддинг L-selectin необходим на более поздних стадиях трансмиграции, чтобы гарантировать регулируемое и поляризованное завершение трансмиграции [81]. На ЭК, экспрессирующих высокие количества VCAM-1, например, при атеросклеротических поражениях, перекатывание моноцитов и переход к медленному перекатыванию и остановке сильно зависят от 1-интегрина VLA-4 [82, 83]. Однако на тромбоцитах, связанных с ЕСМ, скатывание моноцитов скорее зависит от 2-интегрина Mac1 и P-селектина [84].
3.3. Фирменная адгезия
После замедления лейкоциты распознают хемокины, представленные на эндотелии, которые приводят к GPCR-опосредованной передаче сигналов наизнанку, полной активации интегрина и последующей остановке лейкоцитов.Недавно сообщалось, что эндотелиальный рецептор антигена Даффи для хемокинов (DARC) транспортирует CCL2 через эндотелий к апикальной стороне, где он вносит вклад в правильную активацию и рекрутинг моноцитов [85]. В моноцитах полная активация VLA-4 после стимуляции GPCR зависела от сигнальной оси, включая фосфолипазу C (PLC), инозитол-1,4,5-трифосфат (IP3), Ca2 + -флюкс и кальмодулин, но не от PI3K [86], что контрастирует с активацией VLA-4 в нейтрофилах. Хотя каскад адгезии лучше всего изучен на нейтрофилах (обзор в [8]), общим знаменателем, регулирующим переход от перекатывания через медленное перекатывание к остановке во всех лейкоцитах, является активация PLC.Однако, в зависимости от стимула, первичный интегрин, вызывающий прочную адгезию / остановку, варьируется в зависимости от подгруппы лейкоцитов. В случае моноцитов это, по-видимому, VLA-4. Недавно фактор дифференцировки роста-15 (GDF-15) был идентифицирован как эндогенный ингибитор активации VLA-4, который предотвращает связывание моноцитов с VCAM-1 и, таким образом, может служить в качестве местного ингибитора воспаления [87]. Экспрессия VCAM-1 и, следовательно, адгезия моноцитов усиливаются эндотелиальным рецептором протеинтирозинкиназы EphA2 [88, 89].Другими примерами молекул, ингибирующих интегрин эндогенных моноцитов, являются белок эндотелиального матрикса онтогенетический эндотелиальный локус-1 (Del-1) и эндотелиальный CD47, взаимодействующий с сигнальным регуляторным белком моноцитов- α (SIRP-) [90, 91]. Таким образом, моноцит-эндотелиальные взаимодействия, по-видимому, регулируются на разных уровнях, и, скорее всего, в ближайшем будущем будут раскрыты другие механизмы регуляции.
3.4. Ползание
После прочной адгезии лейкоциты распространяются и ползают по эндотелиальной поверхности в поисках подходящего места для трансмиграции.Этот процесс впервые наблюдался с моноцитами и получил название локомоции [92]. Такое направленное движение моноцитов, предшествующее трансмиграции, могло быть заблокировано антителами против LFA-1 или Mac1, что убедительно свидетельствует о зависимости от β 2-интегринов. В случае патрулирования моноцитов антитела против LFA-1, но не против Mac1, отделяли ползающие моноциты, эффект, который также наблюдался у мышей, лишенных CX3CR1 [72]. В нестимулированных венулах кремастера больше моноцитов (по сравнению с нейтрофилами) прилипало и ползало на большие расстояния зависимым от LFA-1 образом.После стимуляции фактором некроза опухоли (TNF-) моноциты сокращали расстояние ползания, которое теперь стало Mac1-зависимым, и большее количество нейтрофилов ползало строго Mac-1-зависимым образом [93]. В этом исследовании блокада Mac1 была более эффективной в снижении экстравазации как моноцитов, так и нейтрофилов по сравнению с блокадой LFA-1. В то время как ползание моноцитов может быть как LFA-1-зависимым, так и Mac1-зависимым, ползание нейтрофилов строго зависит от Mac1 [42], тем самым отмечая разительное различие между этими субпопуляциями лейкоцитов во время экстравазации.
3.5. Стыковочные структуры или трансмиграционные чашки
Во время рекрутирования воспалительных лейкоцитов активированный эндотелий поддерживает нейтрофилы, образуя кластеры вокруг прикрепленных лейкоцитов, которые обогащены LFA-1 / ICAM-1 и VLA-4 / VCAM-1. Эти структуры сначала появляются как кольцеобразные структуры, которые окружают прикрепленные лейкоциты, а затем поглощают лейкоциты в виде стыковочных структур или трансмиграционных чашек, которые обогащены актином и различными адапторными молекулами, такими как кортактин, эзрин, радиксин, моэзин- (ERM-) белки и филамин. и сигнальные молекулы, такие как RhoG и Rac1 [49, 50, 94–100].Другой рецептор эндотелиальной адгезии, обнаруженный в стыковочных структурах, — это активированная молекула адгезии лейкоцитарных клеток-1 (ALCAM-1), которая поддерживает рекрутирование моноцитов в центральную нервную систему (ЦНС) [101]. Док-структуры наблюдались in vitro со всеми лейкоцитами. In vivo подобные структуры, так называемые купола, описаны для нейтрофилов [102]. Для получения дополнительной информации см. Эндотелиальную часть ниже.
3.6. Diapedesis
Чтобы пересечь эндотелиальный монослой между межклеточными соединениями, комплекс VE-кадгерин / катенин должен быть разобран, поскольку он представляет собой физический барьер для трансмиграции лейкоцитов [47].Визуализация трансмиграции in vitro в реальном времени с использованием VE-cadherin-GFP, сверхэкспрессирующего HUVEC, показала, что моноциты, а также нейтрофилы использовали ранее существовавшие и образованные de novo пробелы VE-cadherin для достижения парацеллюлярной трансмиграции [103]. Интересно, что первоначальная трансмиграция моноцитов вызывает подавление VE-кадгерина и активацию PECAM-1, что облегчает последующую трансмиграцию моноцитов [104]. Другие молекулы межэндотелиальных соединений могут фактически использоваться в качестве контррецепторов путем трансмиграции лейкоцитов для облегчения трансмиграции.Например, LFA-1 моноцитов может связываться с JAM-A, а дефицит JAM-A значительно снижает трансмиграцию моноцитов [105, 106]. В головном мозге блокирование взаимодействий JAM-A с LFA-1 снижает трансмиграцию моноцитов и нейтрофилов и улучшает общее неврологическое повреждение после ишемического / реперфузионного повреждения [107]. Экспрессия JAM-подобного белка (JAM-L) активируется на моноцитах во время воспаления и связывается с эндотелиальным рецептором Коксаки и аденовирусным рецептором (CAR), взаимодействие, которое регулируется в cis с помощью VLA-4 [108, 109].Другими молекулами адгезии в контактах ЭК, которые служат противорецептором во время рекрутирования воспалительных моноцитов, являются PECAM-1, CD155 и CD99. PECAM-1 и CD99 взаимодействуют гомофильно, тогда как эндотелиальный CD155 взаимодействует с CD226 на моноцитах. Блокада всех этих молекул, которые являются частью компартмента рециклинга боковой границы (LBRC, [52]), антителами резко снижает TEM моноцитов, не влияя на перекатывание, адгезию и ползание [110–112]. Интересно, что эти молекулы действуют последовательно: сначала происходит вовлечение PECAM-1, затем CD155, а затем CD99 [112].Более того, окклюдин в плотном соединении контролирует трансмиграцию моноцитов через гематоэнцефалический барьер в ответ на метамфетамин зависимым от актина белком-2 / 3- (Arp2 / 3-) [113]. Ингибирование Arp2 / 3 предотвращало индуцированную метамфетамином интернализацию окклюдина и TEM моноцитов.
3,7. События постдиапедеза
Для того, чтобы пересечь BM и слой перицитов, как моноциты, так и нейтрофилы используют области промежутков перицита и низкую экспрессию белков матрикса в BM [10, 69].Однако, в то время как нейтрофилы были способны увеличивать эти области с низкой экспрессией матриксного белка, моноциты проявляли повышенную деформируемость и скорее протискивались через них [69]. NG2 + -перициты также направляют и связывают трансмиграционные моноциты и нейтрофилы за счет секреции хемокинов и экспрессии ICAM-1, соответственно [114]. Общим механизмом для всех лейкоцитов после пересечения слоя ЭК является удлинение уропод и отсроченное отделение лейкоцитов от базальной поверхности эндотелия [68].Этот процесс зависел от взаимодействия LFA-1 с аблюминальным ICAM-1, который поддерживал связь уропода с эндотелием, и, с другой стороны, от VLA-3, который опосредовал движение через BM. Сигналы, необходимые для втягивания хвоста после диапедеза, включают RhoA [115]. Ингибирование RhoA в моноцитах делает клетки неспособными к завершению диапедеза и приводит к накоплению β 2-интегрина в не втянутом хвосте. Напротив, активация RhoA в EC также необходима для эффективной трансмиграции моноцитов [116], которая включает ROCK-опосредованное фосфорилирование легкой цепи миозина и нарушение VE-cadherin-зависимых контактов EC [117].Моноциты, которые полностью трансмигрировали, экспрессируют поверхностный рецептор дискоидинового домена рецептор-1 α (DDR1 α ), который может связывать коллаген и, по-видимому, облегчает миграцию моноцитов внутри богатых коллагеном внеклеточных матриц (ECM) в воспаленных тканях [118]. Интересно, что моноциты могут подвергаться обратной трансмиграции, когда JAM-C-опосредованная адгезия нарушается антителами in vivo [119].
4. Механизмы, используемые лимфоцитами для достижения TEM
Т- и В-лимфоциты (Т- и В-клетки) — это адаптивные иммунные клетки, способные распознавать и различать антигены, что приводит к функциональной специфичности и памяти.Т- и В-клетки возникают из костного мозга и заселяют периферические лимфоидные органы (селезенку и лимфатические узлы), где они завершают созревание и активируются в ответ на специфические антигены, представленные антигенпрезентирующими клетками, включая дендритные клетки (ДК). Перенос в лимфатические узлы происходит посредством L-селектин-зависимой адгезии лимфоцитов к его лигандам на венулах высокого эндотелия (HEV), молекуле-1 адгезии гликан-несущих клеток (GlyCAM-1) и CD34 с последующим интегрин-зависимым арестом через LFA -1 и ВЛА-4.Сродство этих интегринов быстро увеличивается за счет хемокинов CCL19 и CCL21, которые в основном продуцируются HEV в лимфоидных тканях. Эти хемокины затем связываются с CCR7, экспрессируемым как в наивных Т-клетках, так и в В-клетках. После презентации антигена наивные Т- и В-клетки активируются и теряют экспрессию L-селектина [120–122]. Активированные лимфоциты могут стать эффекторными клетками или клетками памяти. Эффекторные В-клетки вырабатывают антитела, которые способствуют уничтожению внеклеточных микробов. Эффекторные Т-клетки могут быть разделены на цитотоксические Т-клетки CD8 + , которые убивают инфицированные вирусом клетки, и подмножества Т-клеток CD4 + , включая Т-хелперные (Th) клетки, которые помогают другим иммунным клеткам выполнять свои функции, и Т-лимфоциты. регуляторные (Treg) клетки, подавляющие функции эффекторных Т-лимфоцитов.Подмножества Т- и В-клеток далее делятся на четко определенные подтипы, которые играют разные роли в здоровье и болезни [123]. В этом разделе мы суммируем различные механизмы, используемые Т- и В-клетками для достижения трансэндотелиальной миграции. Экстравазация субпопуляций Т-клеток была изучена более подробно in vitro и in vivo . В-клетки также инфильтрируют участки воспаления при некоторых заболеваниях [124–126], но предполагается, что В-клетки в значительной степени используют механизмы, аналогичные механизмам Т-клеток.Текущие исследования, посвященные экстравазации В-клеток, обсуждаются в конце этого раздела.
4.1. Механизмы экстравазации Т-клеток
Т-лимфоциты (Т-клетки) — это специализированные лейкоциты, которые обладают способностью распознавать антигены и участвовать в адаптивном иммунном ответе. Процесс рекрутирования Т-клеток в места инфекции или повреждения является фундаментальным в иммунном ответе на экзогенные антигены, что имеет значение для ответов на аутоантигены, которые вызывают хроническое воспаление тканей, когда иммунная толерантность нарушена в аутоиммунитете [127].В рамках каскада рекрутирования Т-клеток адгезия Т-клеток, возможно, является наиболее понятным процессом в условиях потока, с несколькими исследованиями in vitro с участием человеческих CD3 + Т-клеток и линий Т-клеток, характеризующих молекулярные сигналы, регулирующие хемокин- индуцированная активация интегрина [128]. Эти исследования in vitro с использованием человеческих Т-клеток также продемонстрировали, что поведение Т-клеток отличается от других лейкоцитов после установления контакта с активированным эндотелием сосудов.Т-клетки случайным образом прикрепляются к апикальной стороне эндотелия и в течение некоторого времени перемещаются по поверхности. Считается, что это необходимо для того, чтобы дать достаточно времени для индукции перекрестного взаимодействия хемокин-хемокиновый рецептор, необходимого для оптимальной активации интегрина Т-клеток и последующей ТЕА [129]. Это время контакта также позволяет Т-клеточному рецептору (TCR) распознавать потенциальные антигены, представленные сосудистыми ЭК, недавно исследованным путем, который также может приводить к ТЕА [130]. Недавние исследования различных субпопуляций Т-клеток расширили классическую парадигму рекрутирования Т-клеток, выявив новые критические молекулы и пути, ведущие к экстравазации Т-клеток in vivo .Они включают общие, но также и Т-клеточно-специфические молекулы, которые участвуют в различных этапах каскада рекрутирования Т-клеток, как описано ниже.
4.1.1. Rolling
Rolling субпопуляций T-клеток в основном охарактеризован с использованием CD4 + -T клеток, с пониманием того, что многие результаты могут быть верными и для CD8 + -T клеток [131, 132]. Как и в других лейкоцитах, сворачивание Т-клеток происходит в условиях сдвига за счет взаимодействий эндотелиальных селектинов с высокогликозилированными Т-клетками-экспрессируемыми лигандами селектина.В отличие от других лейкоцитов, таких как нейтрофилы и моноциты, которые конститутивно экспрессируют все гликозилтрансферазы, необходимые для биосинтеза функционального селектинового лиганда, они обычно индуцируются и регулируются в Т-клетках [21]. Следовательно, способность Т-клеток связывать эндотелиальные селектины приобретается в ответ на сигналы, которые обеспечивают правильное гликозилирование лигандов селектина. Эти сигналы включают специфические цитокины, и, таким образом, Т-хелперные клетки 1-го типа (Th2) и Т-хелперные 2-го типа (Th3) -клетки, которым требуются разные цитокины для дифференцировки и выживания [133, 134], различаются начальной стадией сворачивания Т-клетки. каскад рекрутирования из-за дифференциальной экспрессии активных лигандов селектина.Клетки Th2 играют важную роль в иммунных ответах на внутриклеточные микробы и в повреждении тканей, связанном с аутоиммунитетом и хроническими инфекциями. Они экспрессируют высокие уровни гликозилтрансфераз в ответ на Th2 цитокин IL-12 и, таким образом, имеют сильно гликозилированные лиганды селектина для катания по активированному эндотелию. Клетки Th3 вносят вклад в борьбу с гельминтными инфекциями и аутоиммунными атопическими заболеваниями, но имеют довольно низкий потенциал экстравазации по сравнению с клетками Th2 [135–137]. Недавно обнаруженные клетки Th27 участвуют в иммунном ответе на внеклеточные бактерии и грибки и, подобно клеткам Th2, играют роль в органоспецифическом аутоиммунитете и хроническом воспалении [138].Интересно, что как Th2, так и Th27 клетки имеют общие лиганды селектина, такие как PSGL-1, которые катятся по эндотелию сосудов посредством P-селектина [139]. Более поздние данные показывают, что эти подмножества, в отличие от Th3 или наивных клеток, могут также использовать Т-клеточный иммуноглобулин и белок муцинового домена 1 (TIM-1) в качестве лиганда P-селектина для опосредования доставки Т-клеток во время воспаления и аутоиммунитета [ 140]. Сходства и различия в механизмах сворачивания наблюдаются во взаимодействиях, опосредованных E-селектин-лиганд / E-селектин среди субпопуляций Т-клеток, при этом клетки Th27 более зависимы от взаимодействий, опосредованных E-селектином, чем клетки Th2 [139].Помимо PSGL-1, который аналогичным образом функционирует в клетках Th2 и Th27 в качестве лиганда как для E-селектина, так и для P-селектина, в клетках Th2 были идентифицированы другие лиганды E-селектина, которые функционируют только во взаимодействии с PSGL-1, и они включают CD44 [141] и CD43 [142, 143]. Дальнейшие исследования определят, могут ли они также функционировать в клетках Th27 специфическим образом.
4.1.2. Адгезия
Новые роли различных субпопуляций Т-клеток в острых и хронических воспалительных процессах и дифференциальная экспрессия хемокиновых рецепторов в различных субпопуляциях Т-клеток недавно были признаны критическими для интегрин-опосредованной адгезии в ответ на специфический хемокин-хемокиновый рецептор. сигнализация [144].Было показано, что связанный с интегрином белок (CD47) регулирует адгезивные функции β 2-интегринов LFA-1 и VLA-4 в Т-клетках in vitro . Было доказано, что это критический механизм, регулирующий адгезию Т-клеток к микрососудам кремастера in vivo в исследованиях, включающих конкурентное привлечение CD47 дикого типа и CD47-дефицитных клеток Th2 [145]. Коактиватор интегрина Kindlin-3, как недавно было показано, усиливает активацию и адгезию интегрина Т-клеток.Интересно, что этот механизм специфичен для адгезии Т-клеток, но не играет роли в диапедезе Т-клеток [146]. Исследования с использованием мышиных клеток Th2 и Th27, генерирующих in vitro , также продемонстрировали, что эти клетки экспрессируют другой репертуар хемокиновых рецепторов. В зависимости от экспонированного хемокинового лиганда эти две подгруппы прикреплялись к иммобилизованному ICAM-1 только в присутствии SDF1 , α и CCL20, соответственно, в условиях сдвигового потока in vitro . Другие исследования с использованием человеческих Т-клеток и HUVEC также продемонстрировали, что CCL20 опосредует адгезию Th27 к ЭК [147].Эти исследования хорошо коррелируют с исследованиями in vivo , показывающими роль CCR6, рецептора CCL20, в специфическом рекрутировании CCR6 + -Th27 клеток в кишечник [148], в центральную нервную систему [149] и кожа [139]. Следовательно, эти механизмы адгезии специфичны для хемокинов / хемокиновых рецепторов. Они также могут быть тканеспецифичными, поскольку клетки Th27 в эндотелии печени используют белок адгезии сосудов-1 (VAP-1) и хемокиновый рецептор CXCR3 в дополнение к CCR6, чтобы опосредовать адгезию через 1 и 2 интегрины [150].
4.1.3. Diapedesis
T-клеточная ТЕА следует за скручиванием и адгезией и в конечном итоге приводит к инфильтрации Т-клеток в воспаленные участки. По сравнению с другими лейкоцитами, такими как нейтрофилы, процент Т-клеток, которые подвергаются ТЕМ в анализах in vitro в условиях потока, намного меньше, и обычно требуются дополнительные хемокины, такие как SDF1, для облегчения остановки Т-клеток и ТЕА. Сходства с другими лейкоцитами включают ICAM-1-опосредованную передачу сигналов при адгезии Т-клеток и пробелы VE-кадгерина в месте трансмиграции соединений [151–153].Недавняя работа продемонстрировала, что Т-клетки могут запускать диссоциацию эндотелиальной рецепторной фосфатазы VE-PTP от VE-кадгерина в качестве механизма, ведущего к фосфорилированию VE-кадгерина и образованию промежутков для облегчения трансмиграции [154]. Эндотелиальный CD47 может также способствовать фосфорилированию VE-кадгерина и участвовать в трансмиграции Т-клеток in vitro . Интересно, что CD47, экспрессируемый на Т-клетках, также необходим для Т-лимфоцитов TEM in vitro и для рекрутирования Т-клеток в участки кожного воспаления in vivo [155]. In vitro , VAP-1 может опосредовать ТЕМ Т-клеток через ЭК печени [156]. Более поздние исследования продемонстрировали, что VAP-1 вместе с общим лимфатическим эндотелиальным и эндотелиальным рецептором сосудов (CLEVER-1) и ICAM-1 могут специфически регулировать TEM Treg-клеток [157]. VAP-1, CLEVER-1 и ICAM-1 высоко экспрессируются в сайтах рекрутирования лейкоцитов в воспаленную печень, что позволяет предположить, что они также регулируют трансмиграцию Т-клеток в печень in vivo . Из всех этих исследований ясно, что хемокины, представленные эндотелием, имеют решающее значение для интегрин-зависимой адгезии и ТЕМ эффекторных Т-клеток и Т-клеток памяти in vitro .Это имеет важные последствия in vivo , где градиенты хемокинов присутствуют в контексте инфекции или повреждения, приводящего к трансмиграции Т-клеток. Новым альтернативным механизмом, описанным для трансмиграции Т-клеток, является способность эффекторных Т-клеток получать доступ не только к внеклеточным депонированным хемокинам, но также к внутриэндотелиальным хемокинам, таким как CCL2, хранящимся в везикулах внутри ЭК, для достижения трансмиграции [158].
Поскольку различные субпопуляции Т-клеток были идентифицированы как основные участники хронического воспаления, роль сосудистого эндотелия рассматривается как критическая для паттернов миграции, приобретаемых опытными антигенами эффекторными Т-клетками, которые мигрируют к участкам хронического воспаления.Остается исследовать, являются ли эти пути специфическими для субпопуляции Т-клеток или специфичными для органа / сосудистого русла / заболевания [164]. Учитывая, что ЭК экспрессируют основные молекулы комплекса гистосовместимости (MHC) I и II и, следовательно, могут функционировать как антигенпредставляющие клетки как для CD4 + -, так и для CD8 + -T-клеток, недавно было признано, что во время контакта и презентация антигена, EC может импринтировать ограниченные, специфические молекулы транспорта в Т-клетках. Считается, что они приобретаются в органе, в котором образовались Т-клетки [165], или в месте инфекции или воспаления, где эти антигенные сигналы, как полагают, способствуют привлечению этих субпопуляций Т-клеток [166, 167].Таким образом, классическая парадигма индуцированной хемокинами остановки Т-клеток и ТЕА в настоящее время подвергается сомнению с помощью альтернативных способов, которые Т-клетки используют для достижения ТЕА при различных воспалительных процессах. In vitro , как эффекторные, так и клетки памяти CD4 + — и CD8 + -T-клетки динамически исследуют эндотелий, расширяя богатые актином инвадосомы / подосомы, подобные выступам (ILP) [168], которые, как считается, активно участвуют в TEM посредством искажая актиновые филаменты и нарушая эндотелиальный барьер [9]. Исследования на мышах in vivo показали, что антигенспецифические CD4 + -эффекторные Т-клетки используют управляемые родственным антигеном сигналы, представленные MHC-II, для проникновения в островки поджелудочной железы при аутоиммунном диабете [169, 170]. Считается, что апикальная презентация родственных антигенных пептидов MHC-I и периваскулярными дендритными клетками увеличивает адгезионную способность интегрина и TEM CD8 + -T-клеток в сосудистых ложах, дефицитных по адгезивной и хемотаксической активности, таких как островки поджелудочной железы при диабете [167] и васкуляризированные трансплантаты [171]. Исследования in vitro в условиях потока с использованием человеческих эффекторных клеток и CD4 + -Т клеток памяти человека способствовали более глубокому пониманию механизмов, имеющих место в ТЕА, опосредованных презентацией антигена и сигналами TCR, по сравнению с классическими индуцированными хемокинами ТЕА, не связанными с антигенными сигналами . Эти исследования продемонстрировали, что Т-клетки могут быстро трансмигрировать в ответ как на хемокины, так и на антигенные сигналы, активирующие TCR, но эти два механизма различаются некоторыми молекулярными путями, регулирующими TEM: TCR-стимулированный TEM сильно зависит от фракталкина (CX3CL1), PECAM- 1, CD99, нектин-2, рецептор полиовируса (CD155) и ICAM-1, тогда как TEM, стимулированный хемокинами, включал ICAM-1 и JAM-A, но не любые другие молекулы [172].Кроме того, оба этих пути ТЕА запускали активацию белка ZAP-70 в трансмиграционной Т-клетке, но различаются по передаче сигналов ниже ZAP-70. Активация Vav-1, Rac-1 и миозина 2A происходила только в Т-клетках, которые контактировали с ЭК сосудов антиген-TCR-зависимым образом [173]. Фенотипически эта передача сигналов приводила к различной реорганизации цитоскелета Т-клеток во время трансмиграции, при этом центр организации микротрубочек Т-клеток (MTOC) организовывался в области контакта между Т-клеткой и EC.Управляемый динеином транспорт гранзим-содержащих гранул в область контакта между Т-клетками и ЭК был идентифицирован как механизм, регулирующий реорганизацию Т-клеточного цитоскелета во время ТЕА [174]. Таким образом, эти специфические молекулярные сигналы, наблюдаемые при управляемой TCR трансмиграции Т-клеток, очень напоминают формирование иммунных синапсов и, по-видимому, являются новым процессом, который Т-клетки используют для достижения успешной ТЕА.
Взятые вместе, Т-клетки уникально специализированы, чтобы реагировать на антигены, пролиферировать и дифференцироваться на подмножества, которые приобретают мигрирующие фенотипы, которые позволяют им перемещаться к участкам воспаления, ранее доступным для нейтрофилов и моноцитов.Т-клетки разделяют некоторые из этих механизмов набора с другими лейкоцитами и запускают аналогичные сигналы в эндотелии сосудов для достижения ТЕА. Специализированный Т-клеточный ответ на различные антигены и цитокиновую среду приводит к отдельной экспрессии активных лигандов селектина и различному репертуару хемокиновых рецепторов, участвующих в свертывании и остановке сосудистого эндотелия. После прикрепления к эндотелию они могут использовать классические пути ТЕА и новые антиген-зависимые пути. Понимание механизмов, которые регулируют рекрутирование эффекторных Т-клеток в различных воспалительных условиях, прольет новый свет на потенциальные способы использования этих путей в иммунотерапевтических целях.
4.2. Механизмы экстравазации В-клеток
Как упоминалось выше, В-клетки в целом используют те же основные механизмы, что и наивные Т-клетки, для размещения вторичных лимфоидных органов. То, как субпопуляции активированных В-клеток мигрируют в определенные ткани во время воспаления, не исследовалось так подробно, как для субпопуляций Т-клеток, для которых был проанализирован каждый этап каскада рекрутирования in vitro и in vivo . Однако некоторые исследования выявили некоторые различия в экстравазации воспалительных В-клеток по сравнению с Т-клетками, которые будут обсуждаться здесь.Многие зрелые В-клетки, называемые плазматическими клетками, мигрируют из лимфатических узлов в костный мозг, где они секретируют антитела IgG в течение длительных периодов времени, которые распределяются по организму через кровоток. Эта субпопуляция В-клеток экспрессирует VLA-4 и CXCR4, которые связываются с VCAM-1 и CXCL12, соответственно, экспрессируемыми в синусоидальных эндотелиальных клетках костного мозга. Напротив, зрелые В-клетки, которые продуцируют антитела IgA, экспрессируют 47, CCR9 и CCR10, которые связываются с MadCAM-1, CCL25 и CCL28, соответственно, экспрессируются в эндотелиальных клетках слизистой оболочки и мигрируют в ткани слизистой оболочки, такие как кишечник [175, 176 ].Считается также, что эти молекулы опосредуют привлечение IgG и IgA-продуцирующих В-клеток к участкам хронического воспаления в синовиальной оболочке [124], головном мозге [125] и стенке сосудов [126]. Более того, CXCL12 был способен стимулировать диапедез В-клеток человека через эндотелиальные клетки микрососудов головного мозга человека в условиях потока, и это блокировалось антителами, блокирующими функцию CXCR4 [159]. Недавно было показано, что ADAM28, который высоко экспрессируется в В-клетках, но не в Т-клетках, связывается с VLA-4 и увеличивает VLA-4-зависимую адгезию линии клеток мышиной B-лимфомы L1-2 к VCAM-1 и последующая трансэндотелиальная миграция предполагает, что эта металлопротеиназа влияет на эффективность экстравазации В-клеток [177].Также было показано, что взаимодействия ephrin-A4 с его эндотелиальным рецептором EphA2 регулируют как нормальную, так и лейкемическую трансэндотелиальную миграцию B-клеток [178]. В-клетки также присутствуют в хронически воспаленной ткани печени. Используя анализ in vitro на адгезию потока и эндотелиальные клетки синусоидальных синусоид печени, B-клетки, полученные из крови человека, были захвачены с помощью VCAM-1 без необходимости в предыдущей фазе вращения и оставались статичными до достижения трансэндотелиальной миграции, опосредованной ICAM-1, VAP-1 и хемокиновые рецепторы CXCR3 и CXCR4.Этот механизм представляет собой заметное различие в экстравазации В-клеток, поскольку Т-клетки демонстрируют энергичное ползание перед трансмиграцией в ту же систему [160]. Другие наблюдали так называемые «интраэндотелиальные канальцевые» структуры, которые особенно используются В-клетками для прохождения через эндотелий во время хоминга и воспаления [179]. В будущем необходимо проанализировать, можно ли наблюдать эти структуры во время экстравазации лейкоцитов других типов.
5. Динамика диапедеза лейкоцитов с точки зрения эндотелия
В спокойных условиях эндотелий экспрессирует низкие уровни молекул адгезии, что позволяет ограниченное иммунное наблюдение.Однако при обнаружении инфекции или повреждения ткани наблюдаемые моноциты и нейтрофилы запускаются для высвобождения воспалительных цитокинов, таких как TNF- α и IL1 β . Важность этого была недавно подчеркнута группой доктора Нуршара [180]. Они показали на элегантной модели in vivo , что нейтрофилы локально секретируют TNF- α , который немедленно действует на эндотелий и тем самым помогает другим нейтрофилам в трансмиграции. При более длительных периодах воспаления ЭК реагируют на медиаторы воспаления путем массивной активации рецепторов адгезии, таких как Р-селектин, Е-селектин, ICAM-1 и VCAM-1, тогда как ICAM-2, как было обнаружено, снижается [181, 182 ].Эти молекулы адгезии привлекают циркулирующие иммунные клетки, чтобы прикрепиться к эндотелиальному монослою и мигрировать через него (рис. 1). Более того, воспалительные цитокины вызывают презентацию хемоаттрактантов на поверхности ЭК, таких как CXCL4 / 5 и IL-8 [14]. Во время сворачивания рецепторы, связанные с G-белком, на лейкоцитах сталкиваются с такими хемокинами, представленными ЕС, и передают сигнал, чтобы вызвать конформационное изменение интегринов лейкоцитов LFA-1 ( α L β 2 ) и VLA-4 ( α 4 β 1 ) в состояние высокого сродства, позволяющее этим интегринам взаимодействовать со своими эндотелиальными лигандами ICAM-1 и VCAM-1, соответственно [13, 183].Как следствие, прочная адгезия, ползание и, наконец, диапедез происходят через межэндотелиальные соединения или через тело ЭК [184]. Хотя считается, что эти процессы происходят для любого типа лейкоцитов, пересекающих эндотелий, некоторые эндотелиальные сигналы специфически индуцируются определенными подмножествами лейкоцитов, которые мы выделим в этой главе.
5.1. Повышение регуляции молекулы адгезии
Воспалительный цитокин TNF- стимулирует ЭК, связываясь с его рецептором TNFR1 (CD120a) [185, 186].Это связывание индуцирует ассоциацию цитоплазматического адаптивного белка TRADD (TNFR1-ассоциированный белок домена смерти) с внутриклеточным доменом TNFR1. Впоследствии TRADD связывается с нижестоящими эффекторами, такими как серин / треонинкиназа RIP1 (белок 1, взаимодействующий с рецептором), а также с E3-убиквитинлигазой TRAF2 (фактор 2, связанный с TNFR). Эта ассоциация, в свою очередь, запускает каскад передачи сигналов киназ, ведущий к активации митоген-активируемых протеинкиназ (MAPKs) p38, JNK и ERK [187].Эти киназы способны активировать факторы транскрипции, такие как протеин-активатор-1 (AP-1). Кроме того, TRAF2 и RIP1 индуцируют активацию фактора транскрипции NF-B. В условиях покоя NF-B удерживается в цитозоле ингибитором B (IB). При активации комплекса киназы IB (IKK) с помощью TRAF2 и RIP1, IB фосфорилируется, что приводит к его деградации и последующей ядерной транслокации NF-B [188].
Промоторы молекул адгезии E-селектин, VCAM-1 и ICAM-1 содержат несколько сайтов связывания NF-B, и было показано, что NF-B является основным регулятором экспрессии молекул адгезии, индуцированной TNF в EC [189–194].Хотя промоторы E-selectin, VCAM-1 и ICAM-1 также содержат AP-1-связывающие мотивы, эти сайты вносят различный вклад в индуцированную TNF активацию этих молекул адгезии [192, 193, 195, 196]. Кроме того, известно, что другие факторы транскрипции, такие как регуляторный фактор-1 интерферона (IRF-1), специфический белок 1 (Sp1) и GATA, активируются посредством плохо охарактеризованных сигнальных путей и способствуют адгезии, индуцированной TNF- α . активация молекулы в ЭК [191, 197–199].
5.2. Передача сигналов с помощью САМ
Паттерны экспрессии интегрина различаются в зависимости от типа лейкоцитов. Например, нейтрофилы в первую очередь экспрессируют Mac1 и LFA-1 и почти не экспрессируют VLA-4, тогда как моноциты, а также Т-лимфоциты и дендритные клетки экспрессируют все три интегрина, хотя и на разных уровнях [200]. Это уже указывает на то, что разные типы лейкоцитов через свой репертуар интегринов могут кластеризовать разные лиганды на эндотелии, что приводит к отличительным внутриклеточным сигналам в эндотелии, которые различаются в зависимости от типа лейкоцитов.Например, Th27-лимфоциты показали повышенную адгезию к E-селектину по сравнению с Th2-лимфоцитами, скорее всего, из-за лучшей активации интегрина на этих клетках через ось CCL20-CCR6 [139].
ICAM-1 и VCAM-1 являются членами суперсемейства иммуноглобулинов (Ig) молекул адгезии, внеклеточные домены которых характеризуются наличием пяти и шести Ig-подобных доменов, соответственно. По сравнению с их эктодоменами, ICAM-1 и VCAM-1 имеют относительно небольшие карбоксил- (C-) концевые внутриклеточные домены, состоящие всего из 28 и 19 аминокислот соответственно.Хотя C-концевые домены не содержат каких-либо явных сигнальных мотивов, внутриклеточный домен ICAM-1, как было показано, необходим для эффективного TEM лейкоцитов [201, 202]. Более того, вовлечение ICAM-1 с помощью LFA-1 / Mac1 было связано с реорганизацией F-actin и с инициацией сигнальных событий внутри EC [203]. Несколько исследований показали, что адгезия лейкоцитов и кластеризация ICAM-1 вызывают повышение внутриклеточных уровней Ca 2+ [204, 205], что приводит к активации тирозинкиназы Src протеинкиназой C (PKC).В свою очередь, Src индуцировал фосфорилирование тирозина белков фокальной адгезии, таких как паксиллин, кортактин и FAK [204, 206, 207]. Кластеризация ICAM-1 ведет к активации небольшой RhoGTPase RhoA, которая стимулирует образование стрессовых волокон F-actin и увеличивает проницаемость эндотелиального монослоя [204, 208–210] (Figure 2). Более того, было продемонстрировано, что активность RhoA также необходима для эффективного рекрутирования ICAM-1 вокруг прикрепившихся моноцитов [210], указывая на вышестоящую роль RhoA в индуцированном ICAM-1 сигнальном каскаде.Недавно было показано, что кластеризация ICAM-1 индуцирует фосфорилирование тирозина VE-кадгерина Src- и Pyk2-зависимым образом, что совпадает с повышенной проницаемостью эндотелия [203, 211, 212]. Мартинелли и его коллеги показали, что кластеризация ICAM-1 индуцирует фосфорилирование eNOS на S1177, и это регулируется киназой Src, а также киназой RhoA, кальция, CaMKK и AMP, но не киназой PI3. Они дополнительно показали, что этот путь контролирует фосфорилирование VE-кадгерина и перенос лимфоцитов [203].В отличие от ICAM-1, только несколько исследований сообщили о сигнальных событиях, индуцированных при взаимодействии и кластеризации VCAM-1. Интегрин лейкоцитов VLA-4, экспрессируемый на моноцитах и лимфоцитах, показал сильное предпочтение связывания с VCAM-1 [213]. Кластеризация VCAM-1, как было показано, способствует активации Rac1, ведущей к продукции активных форм кислорода (ROS) [214–216]. Было продемонстрировано, что VCAM-1-зависимая продукция ROS регулирует активацию матриксных металлопротеиназ, которые могут вносить вклад в локальное разрушение соединений эндотелиальных адгезивов [217].Кроме того, было показано, что кластеризация VCAM-1 регулирует TEM лимфоцитов путем активации киназы PKC α и тирозинфосфатазы PTP1B ROS-зависимым образом [218, 219].
В дополнение к классическим молекулам адгезии на эндотелии (например, ICAM-1/2 и VCAM-1) известно, что несколько других молекул играют важную роль в трафике лейкоцитов. Некоторые из них относятся к эктоферментам, которые представляют собой молекулы клеточной поверхности, имеющие каталитически активные центры вне клетки.Например, было обнаружено, что молекула адгезии Vascular Adhesion Molecule-1 (VAP-1) с ферментативной активностью аминоксидазы присутствует на поверхности эндотелия и контролирует движение лимфоцитов [220–222], моноцитов [161] и нейтрофилов [ 162, 163]. Однако неизвестно, передают ли эти эктоферменты внутриклеточные сигналы в эндотелий, которые ремоделируют актиновый цитоскелет во время ТЕА лейкоцитов.
Трансмембранный белок CD47 также является важным медиатором переноса лейкоцитов [145, 155].CD47 экспрессируется на многих, если не на всех типах лейкоцитов, а также на EC и взаимодействует с SIRP γ , который экспрессируется на лимфоцитах [223]. Та же группа показала, что CD47 может фосфорилировать VE-кадгерин и, таким образом, опосредовать TEM лимфоцитов, снова Src- и Pyk2-зависимым образом [155]. Интересно, что перекрестное сшивание CD47 с антителами приводит к образованию стрессовых волокон, аналогично тому, что наблюдалось при перекрестном связывании ICAM-1 [209, 210, 224]. Очевидно, что изменения в актиновом цитоскелете эндотелия, вызванные связыванием лейкоцитов, эффективно контролируют ТЕА лейкоцитов.
Тетраспанины образуют микродомены в плазматической мембране и участвуют в межклеточной адгезии и миграции. Для TEM лимфоцитов и моноцитов сообщалось, что тетраспанины CD9, CD81 и CD151 распределяются по сайту контакта с трансмиграционными лейкоцитами и латерально связываются как с ICAM-1, так и с VCAM-1 [225, 226]. Они контролируют адгезионную способность молекул адгезии и тем самым контролируют силу связывания лейкоцитов с эндотелием. Кроме того, Barreiro и соавторы обнаружили, что тетраспанины могут образовывать так называемые эндотелиальные адгезивные платформы (EAP), с которыми лейкоциты могут связываться [95].Эти платформы могут функционировать как центры передачи сигналов в плазматической мембране и могут также включать липидные рафты. Интересно, что и ICAM-1, и VCAM-1 могут присутствовать на этих платформах независимо от присутствия их рецептора.
Сводка описанных выше сигнальных путей ниже кластеризованных ICAM-1 и VCAM-1 показана на рисунке 2, где мы обозначили цветом эндотелиальные белки, которые активируются определенными подмножествами лейкоцитов.
5.3. Связывание CAM с цитоскелетом F-Actin
Чтобы поддерживать надлежащую адгезию в условиях физиологического потока, ICAM-1 и VCAM-1 должны быть внутриклеточно прикреплены к цитоскелету.В последние два десятилетия сообщалось, что несколько адаптерных белков актина взаимодействуют с внутриклеточными доменами VCAM-1 и ICAM-1. Эти адаптерные белки связывают эти молекулы с цитоскелетом F-актина (рис. 2). Было обнаружено, что адаптерные белки эзрин и моэзин из семейства ERM напрямую взаимодействуют с VCAM-1. Более того, они колокализовались с VCAM-1 вокруг прикрепленных лимфобластов [94]. Их способность связывать как фосфолипиды, так и F-актин позволяет белкам ERM организовывать молекулы адгезии в специализированные мембранные домены [227].Помимо VCAM-1, белки ERM, как сообщается, также взаимодействуют с ICAM-1 зависимым от PIP 2 образом и совместно локализуются с ICAM-1 в микроворсинчатых структурах [94, 210, 228, 229]. Однако, в отличие от связывания с VCAM-1, взаимодействие эзрина и моэзина с ICAM-1, как сообщается, является непрямым [230]. В дополнение к ERM белкам, связывающие F-актин белки , α, -актинин-1 и -4, также продемонстрировали взаимодействие с C-концом ICAM-1 через кластер ICAM-1-C-концевых положительно заряженных аминокислот. [231, 232].Интересно, что этот же кластер аминокислот, как было показано, опосредует взаимодействие ICAM-1 с эзрином [229], предполагая, что α -актинин и белки ERM могут конкурировать за связывание с ICAM-1. Это также указывает на существование различных комплексов ICAM-1 / актин при кластеризации, опосредованной лейкоцитами (Рис. 2).
Первоначально было показано, что кортикальный актин-связывающий белок кортактин становится фосфорилированным по тирозину после кластеризации ICAM-1 [206], и это фосфорилирование тирозина необходимо для эффективного ТЕМ нейтрофилов [100].Считается, что кортактин стабилизирует разветвленные актиновые сети посредством взаимодействия с комплексом Arp2 / 3 [233]. Он также связан с ICAM-1 при кластеризации [234] и необходим для рекрутирования ICAM-1 и F-актина в кольцеобразные структуры вокруг прикрепленных лейкоцитов [99]. Недавно было показано, что кортактин также необходим для кластеризации ICAM-1 вокруг прикрепившихся нейтрофилов и для эффективной экстравазации нейтрофилов in vivo , таким образом подчеркивая физиологическое значение взаимодействия ICAM-1-кортактин [49].
Наконец, Кантерс и его коллеги показали, что белок, перекрестно сшивающий F-actin, filamin B, взаимодействует с C-концом ICAM-1 прямым образом [235]. Подобно кортактину, филамин B требовался для привлечения ICAM-1 к кольцу вокруг прикрепившихся нейтрофилов и для ТЕА нейтрофилов в условиях физиологического потока. В более поздней публикации было показано, что филамин А также взаимодействует с внутриклеточным хвостом ICAM-1 [236]. Поэтому было высказано предположение, что эти адаптерные белки соединяют ICAM-1 с нижестоящими партнерами по передаче сигналов [237, 238].Подавление экспрессии филамина B нарушало кластеризацию ICAM-1 и ТЕМ лейкоцитов в физиологических условиях потока, и, поскольку филамин A все еще присутствовал в EC с подавленным филамином B, это предполагает, что филамины не являются функционально избыточными.
В самом деле, хотя филамины A и B имеют 70% идентичности аминокислотных последовательностей, наблюдались различные эффекты на функцию ICAM-1, когда либо филамин A, либо филамин B подавлялись. Используя технологию восстановления флуоресценции после фотообесцвечивания (FRAP), стало ясно, что подавление экспрессии филамина B увеличивает неподвижную фракцию ICAM-1 в плазматической мембране [235].Напротив, в ЭК без филамина А неподвижная фракция ICAM-1 была снижена [239]. Кроме того, индуцированная кластеризацией ассоциация ICAM-1-актин была нарушена в EC с подавленным филамином A, но не в EC с подавленным филамином B (личное наблюдение, JDvB). Эффекты дефицита филамина А на функцию ICAM-1 аналогичны эффектам ингибирования полимеризации F-актина. Более того, делеция внутриклеточного домена ICAM-1 снижает неподвижную фракцию ICAM-1 [239]. Кроме того, филамин A, но не филамин B, также опосредует взаимодействие ICAM-1 с маркером липидного рафта и основным компонентом кавеол, кавеолином-1 [235].Поскольку ICAM-1 рекрутируется в кавеолы и кавеолин-1 во время TEM трансцеллюлярных лимфоцитов [240], filamin A может играть специфическую роль в регуляции трансцеллюлярного пути диапедеза. Таким образом, эти находки показывают важную роль различных филаминов в контроле динамики ICAM-1 путем регулирования связи с цитоскелетом F-actin и специфическими мембранными доменами.
Недавно стало ясно, что существует иерархия между этими актин-связывающими белками, которые связываются с ICAM-1 при кластеризации.Шефер и его коллеги показали, что при кластеризации ICAM-1 α -актинин является первым белком, который будет задействован в ICAM-1, за ним следуют корактин и, наконец, филамин [241]. Привлечение разных адаптерных белков к ICAM-1 может приводить к составу другой актиновой сети. Например, α -актинин перекрестно связывает актиновые филаменты в актиновые пучки, тогда как кортактин перекрестно связывает актин в сетку, а филамин в «гелеобразную» структуру [233, 237]. Инициирование этих различных сетей актина может генерировать силы, которые управляют локальной протрузивной активностью, то есть стыковкой структур, или создают поверхность, по которой лейкоциты могут ползать.Фактически, они показали, что локальная жесткость ЭК действительно зависит от α -актинина. Истощение α -актинина привело к снижению способности нейтрофилов распространяться и трансмиграции [241]. Группа д-ра Кармана недавно показала, что морфология цитоскелета и, как следствие, локальная жесткость ЭК различных сосудистых слоев определяют предпочтительный путь прохождения Т-лимфоцитами эндотелия [9]. В частности, высокая барьерная функция была связана с трансцеллюлярной миграцией, тогда как искусственное открытие соединений приводило к большей параклеточной миграции.Ранее они показали, что Т-лимфоциты используют инвадоподии-подобные выпячивания для исследования эндотелиальной поверхности, возможно, для инициации трансцеллюлярной миграции [242]. Возникает соблазн предположить, что скорость кластеризации молекул адгезии, таких как ICAM-1 или VCAM-1, определяет жесткость подлежащей эндотелиальной поверхности и что это может быть спусковым крючком, по крайней мере, для пересечения Т-лимфоцитов. Неизвестно, используют ли другие типы лейкоцитов, например нейтрофилы или моноциты, тот же механизм для исследования поверхности.
Недавнее исследование группы Woodfin показало, что, наряду с ICAM-1 и VCAM-1, ICAM-2 играет важную роль в трафике иммунных клеток in vivo , как описано выше [45]. Неясно, привлекает ли кластеризация ICAM-2 адаптерные белки актина и индуцирует ли сходные сигналы [202]. Однако роль ICAM-2, по-видимому, больше ограничена определенными органами. Например, эндотелиальный ICAM-2 необходим для миграции Т-клеток через гематоэнцефалический барьер [243, 244].
5.4. Формирование эндотелиальной стыковочной структуры
Используя конфокальную микроскопию, Баррейро и его коллеги показали, что как ICAM-1, так и VCAM-1 были задействованы в выступах мембран, богатых актином, которые окружают прилипшие Т-лимфобласты в чашевидных структурах, которые были названы эндотелиальными стыковочными структурами. [94]. Два последующих исследования Carman с соавторами продемонстрировали, что образование этих структур зависело от полимеризации F-актина и сильно коррелировало с трансмиграцией лейкоцитов [96, 97].Они предположили, что эти структуры могут функционировать, облегчая и направляя ТЕМ лейкоцитов, формируя чашевидную структуру тяги, которая выровнена параллельно направлению трансмиграции. Интересно, что они показали, что трансмиграционные чашки практически равны между моноцитами, нейтрофилами и лимфоцитами. Таким образом, чашки не различали типы лейкоцитов, что позволяет предположить, что эти эндотелиальные чашки представляют собой более глобальный механизм экстравазации лейкоцитов. В отличие от того, что предложили Баррейро и его коллеги, то есть, что стыковочные структуры необходимы для адгезии лейкоцитов, Карман и его коллеги показали, что чашки были сильно связаны с лейкоцитами, которые мигрировали.Разрушение чашечек не повлияло на способность лейкоцитов прилипать к эндотелию даже в условиях потока. Интересно, что образование чашечек зависело от внутриклеточного хвоста ICAM-1 [98]. В соответствии с представлением о том, что чашки не участвуют в адгезии, несколько отчетов показали, что внутриклеточный хвост ICAM-1 необходим для правильного диапедеза, но не для прочной адгезии [201, 202, 229]. В дополнение к наблюдениям in vitro , многочисленные исследования также описали формирование эндотелиальных стыковочных структур in vivo [102, 245–249].Таким образом, хотя определенные доказательства все еще отсутствуют, образование стыковочных структур показывает сильную корреляцию с этапом диапедеза.
Начальное формирование эндотелиальных стыковочных структур зависит от активности малой GTPase RhoG [238]. RhoG колокализовался с ICAM-1 после кластеризации ICAM-1 и был активирован. Более того, истощение SGEF, GEF для RhoG или RhoG значительно снижает образование стыковочных структур и ТЕМ нейтрофилов. Используя мышиную модель для изучения формирования атеросклероза, стало ясно, что SGEF, скорее всего, участвует в привлечении моноцитов к месту повреждения, поскольку животные с дефицитом SGEF показали значительное уменьшение бляшек по сравнению с контрольными животными [250].В этой работе подчеркивается важность стыковки структур в развитии воспалительных заболеваний, таких как атеросклероз.
Интересно, что Дуле и его коллеги показали, что сигнальные молекулы, которые обычно отвечают за индукцию структур апикальной чашечки, могут использоваться бактериями (например, Neisseria meningitides ) для проникновения в ЭК [251]. Эти бактерии титровали адаптерный белок актина эзрин и моэзин вдали от участков, где лейкоциты взаимодействовали с эндотелием, и тем самым предотвращали образование чашеобразных структур и диапедез лейкоцитов.Это исследование показывает потенциальную клиническую значимость чашеобразных структур в ТЕА лейкоцитов во время воспаления. То, как эти патогены проникают через стенку сосуда и проникают в клетки-хозяева, может многое рассказать нам об основных принципах сигнальных механизмов во время ПЭМ лейкоцитов.
6. Выводы
Первоначальная многоэтапная парадигма экстравазации лейкоцитов в значительной степени описывает адгезию и диапедез с точки зрения лейкоцитов и рассматривает эндотелий просто как пассивный субстрат для адгезии лейкоцитов.Однако сейчас хорошо известно, что эндотелий также является активным участником этого процесса. Было продемонстрировано, что кластеризация молекул адгезии, таких как ICAM-1 и VCAM-1, индуцирует передачу сигналов, приводящую к значительным изменениям в морфологии ЭК, что делает возможным пассаж лейкоцитов. Кроме того, образуются структуры эндотелиальных чашечек, которые могут захватывать лейкоциты и направлять их для их миграции через эндотелий. Таким образом, существенная роль рецепторов адгезии эндотелия и актин-связывающих белков в обеспечении ТЕА лейкоцитов делает их многообещающими кандидатами для целенаправленной регуляции экстравазации лейкоцитов.С другой стороны, в лейкоцитах было идентифицировано несколько механизмов, которые активируют, например, интегрины для правильного взаимодействия с EC и динамикой актина, вызывая необходимые морфологические изменения во время TEM. Некоторые из таких механизмов были идентифицированы во всех подгруппах лейкоцитов, в то время как другие, по-видимому, специфичны для данной подгруппы. Тем не менее, являются ли они действительно специфическими или просто еще не исследованы в других подмножествах, еще предстоит выяснить для большинства описанных механизмов. Важно помнить, что все типы лейкоцитов, помимо их способности к разрушению тканей, выполняют полезные функции во время многих патофизиологических состояний, так что фармакологическое воздействие на рекрутинг лейкоцитов, скорее всего, всегда будет иметь положительные и вредные эффекты.Таким образом, предстоит проделать большую работу, пока мы не сможем полностью оценить, существуют ли действительно уникальные механизмы, используемые различными подгруппами лейкоцитов во время ТЕА, на которые можно было бы воздействовать фармакологически при определенных патологических состояниях, которые выиграют от вмешательства в набор только одного данного лейкоцита. тип, не затрагивая других.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов в отношении публикации данной статьи.
Вклад авторов
Все авторы внесли равный вклад в эту работу.
Благодарности
Работа в лаборатории Майкла Шнора финансируется за счет грантов Мексиканского совета по науке и технологиям (Conacyt: 179895, 207268 и 233395). Работа в лаборатории Пилар Алкайд финансируется грантами Американской кардиологической ассоциации (AHA GIA 13GRNT 14560068) и Национальных институтов здоровья (NIH HL097406 и HL123658). Работа в лаборатории Матье-Бенуа Вуазена финансируется компанией Arthritis Research UK (19913).Работа в лаборатории Яапа Д. ван Буула финансируется за счет грантов Голландского фонда сердца (2005T039) и фонда LSBR (1701).
|
Воспаленные нейтрофилы, секвестрированные в захваченных опухолевых клетках посредством хемотаксического удержания, способствуют экстравазации опухолевых клеток
Значимость
Системное воспаление, возникающее во время лечения опухоли, часто коррелирует с неблагоприятными онкологическими исходами независимо от инфекционного инсульта.В качестве первой линии защиты от инфекции нейтрофилы, вероятно, играют важную роль в модулировании прогрессирования отдаленных метастазов в этих сценариях. В этом исследовании используется мультиплексная микрофлюидная модель микроциркуляции человека в сочетании с LPS-стимулированными нейтрофилами в качестве модели системной инфекции для исследования динамических взаимодействий между внутрисосудистыми опухолевыми клетками и нейтрофилами с высоким пространственно-временным разрешением. Мы раскрываем хемокин-зависимые модели миграции нейтрофилов, которые приводят к увеличению скорости экстравазации опухолевых клеток.Эти данные могут служить основой для подавления проэкстравазации и, возможно, прометастатического эффекта воспаленных нейтрофилов.
Abstract
Системное воспаление, возникающее в процессе прогрессирования опухоли и лечения, часто коррелирует с неблагоприятными онкологическими исходами. Таким образом, предполагается, что нейтрофилы, первая линия защиты от инфекции, могут играть важную роль в связывании воспаления и метастатического посева. Чтобы расшифровать динамическую роль воспаленных нейтрофилов во время гематогенного распространения, мы используем мультиплексную микрофлюидную модель микроциркуляторного русла человека, обеспечивающую физиологически значимый транспорт циркулирующих клеток в сочетании с наблюдением в реальном времени гетеротипных межклеточных взаимодействий с высоким пространственным разрешением.LPS-стимулированные нейтрофилы (PMN) и опухолевые клетки (TC) образуют гетеротипические агрегаты в потоке и задерживаются как из-за механического захвата, так и из-за адгезии нейтрофилов к эндотелию. Удивительно, но PMN не являются статичными после агрегации, а демонстрируют ограниченную картину миграции около кластеров TC – PMN. Мы обнаружили, что PMN хемотаксически ограничены самосекретируемым IL-8 и происходящим из опухоли CXCL-1, которые иммобилизованы эндотелиальным гликокаликсом. Это приводит к значительной секвестрации нейтрофилов с задержанными опухолевыми клетками, что приводит к пространственной локализации полученного из нейтрофилов IL-8, что также способствует увеличению потенциала экстравазации соседних опухолевых клеток за счет модуляции эндотелиального барьера.Поразительно похожие паттерны миграции и экстравазация поведения наблюдались также на модели рыбок данио in vivo при совместной инъекции PMN-опухолевых клеток в сосудистую сеть эмбриона. Эти сведения о временной динамике внутрисосудистых взаимодействий опухоль-PMN проливают свет на механизмы, посредством которых воспаленные нейтрофилы могут оказывать проэкстравазационные эффекты в удаленном месте метастазирования.
Для успешного метастазирования опухолевые клетки (ОК) должны претерпевать гематогенное распространение и экстравазацию, процесс, который, как сейчас широко считается, включает как кооперативные, так и антагонистические взаимодействия между факторами микроокружения хозяина и опухолевыми клетками (1).В частности, опухолевые клетки сталкиваются с множеством циркулирующих кровяных клеток, включая нейтрофилы — преобладающие циркулирующие гранулоциты у людей (2). В настоящее время роль нейтрофилов в посеве метастазов является спорной (3). Клинические исследования показали, что нейтрофилия часто предсказывает ухудшение выживаемости, связанной с метастазами (4). В подтверждение этого, нейтрофилы могут усиливать образование метастатических очагов в экспериментальных моделях мышей, что видно по уменьшению метастатической нагрузки при их истощении с помощью анти-Ly6G (5⇓-7) или увеличивается при добавлении экзогенных нейтрофилов человека (8, 9). .Были предложены различные механизмы этого прометастатического эффекта, включая увеличение удержания опухолевых клеток в сосудистой сети за счет адгезии опухоль-нейтрофилы через интегрины (8). Считается, что производные нейтрофилов факторы, такие как LTB4, усиливают колонизацию путем выборочного увеличения подпула раковых клеток с высоким канцерогенным потенциалом (5). Напротив, накопление нейтрофилов в предметастатическом легком может ингибировать метастатическое засевание за счет образования H 2 O 2 , эффекта, опосредованного секретируемым опухолью CCL2 (10, 11).Таким образом, взаимодействия нейтрофилов и опухолевых клеток не могут быть равномерно прометастатическими.
Большое внимание было уделено связи между системным воспалением и метастатическим посевом. В частности, курсы лечения опухолей часто сопровождаются воспалительной активацией, которая коррелирует с неблагоприятными метастатическими исходами, независимо от заболеваемости, связанной с самой воспалительной реакцией (12–14). Воспаление, связанное с лечением, может возникать в результате периоперационных инфекций (12, 15) и других лечебных вмешательств, включая паллиативную хирургию и химиотерапию (стерильное воспаление) (14, 16–19).Нейтрофилы, как правило, играют ключевую роль в воспалительных реакциях, их роль изучается на животных моделях повреждений, включая перевязку слепой кишки и реперфузию ишемии, где образование внеклеточных ловушек нейтрофилов (NET) может задерживать циркулирующие опухолевые клетки и усиливать метастазирование (18, 20). Точно так же предварительная стимуляция нейтрофилов липополисахаридом (ЛПС) увеличивала задержку опухолевых клеток в синусоидах печени и макроскопических метастазах (9). В сочетании с открытием того факта, что после резекции опухоли раковые клетки часто попадают в кровоток (21, 22), очень важно выяснить роль нейтрофилов в связи между воспалением и отдаленными метастазами.
Используя in vitro модель микрососудов человека и LPS-стимулированных нейтрофилов для имитации воспаленного состояния, мы показываем, что начальная внутрипросветная кластеризация TC и PMN зависит как от физического захвата, так и от адгезивных взаимодействий между PMN и эндотелиальным ICAM-1. Однако после ареста PMN внутри кластеров сильно мигрируют, не поддерживая прочных контактов с TC. Интересно, что эти PMN мигрируют ограниченным образом, опосредованным самосекретируемым PMN IL-8 и происходящим из опухоли CXCL-1, заставляя их секвестрироваться в кластеры TC-PMN.Более того, ограничение миграции усиливается гликокаликсом эндотелиальных клеток (ЭК), вероятно, посредством иммобилизации хемокинов. Поразительно, что сходные паттерны ограниченной миграции наблюдались в сосудистой сети модели метастазов у рыбок данио in vivo. Эта секвестрация PMN в непосредственной близости от кластеров опухолевых клеток приводит к пространственной локализации и секвестрации производного PMN IL-8, что приводит к разрушению эндотелиального барьера и усилению экстравазации соседних захваченных опухолевых клеток.
Результаты
Внутрисосудистая агрегация TC – PMN модулируется механическим захватом и адгезивным взаимодействием.
Ранее мы описали и охарактеризовали микрожидкостный анализ in vitro с использованием трехмерных перфузионных микрососудистых сетей для изучения динамики экстравазации опухолевых клеток (23–25). Подробные сведения о методе формирования сосудистой сети и перфузии опухолевых клеток можно найти в другом месте (26). Микроустройство включает самоорганизованные микрососудистые сети человека, образованные эндотелиальными клетками пупочной вены человека (HUVEC) в фибриновых гелях, через которые можно перфузировать ОК и отслеживать события экстравазации с помощью стандартной конфокальной микроскопии.Здесь мы улучшаем существующий анализ с точки зрения пропускной способности, устойчивости формирования перфузируемой сети и простоты использования (Рис. 1 A и SI Приложение , Рис. S1). После формирования сети непрерывный поток может быть установлен через интегрированный резервуар, который применяет пассивный перепад гидростатического давления (Рис. 1 B ).
Рис. 1.Мультиплексный анализ микрососудистой сети позволяет количественно оценить динамику остановки и экстравазации TC – PMN. ( A ) Восемь независимых областей гидрогеля, где микрососудистые сети, соединенные разветвляющимися каналами, образуются на каждом чипе.Флуоресценция На вставке изображена конфокальная проекция одной перфузируемой сосудистой сети. (Шкала: 200 мкм.) ( B ) Резервуар, выдерживающий перепад гидростатического давления ~ 5 мм водяного столба, закреплен на вершине чипа для создания непрерывной перфузии. ( C ) Кластеры TC – PMN в микрососудах. Примеры с большим увеличением экстравазированных и неэкстравазированных клеток MA2 в кластерах TC – PMN. ( D ) Степень образования агрегатов опухолевых клеток и PMN во время внутрисосудистой остановки.Индекс агрегации определяется как доля ОХ, сгруппированных с нейтрофилами, умноженная на среднее количество нейтрофилов на кластер ( n = 10–13 устройств на одно условие). ** P <0,01, *** P <0,001, а полосы ошибок указывают SD.
Чтобы воспроизвести воспаленный фенотип, нейтрофилы стимулировали LPS в течение 30 минут, промывали три раза с последующей коперфузией с клетками меланомы A375-MA2 в сосудистые русла in vitro. После нескольких минут потока многие клетки агрегировались в гетеротипные кластеры (рис.1 C и SI Приложение , рис.2 A ). Предварительная стимуляция LPS активировала экспрессию поверхностного CD11b и впоследствии увеличивала индекс агрегации PMN с TC (рис. 1 D и SI, приложение , рис. S2). Это также привело к большим кластерам и большему количеству PMN, арестованных на устройство, в то время как анти-CD11b ослаблял эту кластеризацию ( SI Приложение , рис. S2 C ). Эти результаты предполагают наличие адгезивных взаимодействий, модулирующих агрегацию TC – PMN.Обработка сосудов или ТК одним анти-ICAM-1 приводила к меньшей кластеризации, при этом блокирование только EC-ICAM-1 давало более сильное снижение. Интересно, что экспрессия ICAM-1 увеличивалась на эндотелиальных клетках только после перфузии клеток MA2, и тем более при MA2 и стимулировании коперфузии PMN ( SI Приложение , рис. S3 A ). Когда опухолевые клетки были заменены шариками размером 15 мкм, интенсивность ICAM-1 не увеличивалась, в то время как агрегация TC – PMN снижалась, что позволяет предположить, что начальная стимуляция эндотелия опухолевыми клетками модулирует адгезию CD11b – ICAM-1 (рис.1 D ). Несмотря на инертную природу гранул, исходный уровень агрегации указывает на то, что механический захват также играет значительную роль в кластеризации. Вопреки нашим ожиданиям, добавление PMN не увеличивало начальную скорость захвата (<5 мин) TC; тем не менее, значительные различия были замечены в удержании ОК (> 3 ч), что свидетельствует о том, что PMN могут повышать устойчивость ОК к сдвиговому потоку ( SI Приложение , рис. S3 B ), увеличивая абсолютное количество клеток, доступных для экстравазии.В целом, наши данные предполагают, что степень начальной кластеризации TC-PMN и общее удержание PMN в сосудистой сети зависят от адгезионных взаимодействий между CD11b нейтрофилов и эндотелиального ICAM-1, активируемых при контакте TC и PMN, в дополнение к физическим механизмам захвата. . Гетеротипическая кластеризация дополнительно предотвращает отток опухолевых клеток.
Кластерные PMN хемотактически ограничены производным от PMN IL-8 и производным TC CXCL-1.
Покадровая съемка отдельных «кластеров TC – PMN» показала, что кластеры PMN не были статичными, а сильно мигрировали по поверхности эндотелия, при этом некоторые из них переходили в окружающую матрицу (фильмы S1 и S2).PMN были разделены на две субпопуляции — «связанные с кластером» или «свободные» — в зависимости от их близости к арестованным кластерам (Рис. 2 A и SI Приложение , Рис. S4 A и B ). Связанные с кластером PMN продемонстрировали как сниженные скорости миграции, так и сквозные смещения по сравнению со свободными PMN в течение 90 минут (рис. 2 B ). Поразительно, что связанные с кластером PMN оставались более ограниченными своим исходным положением, тогда как свободные PMN демонстрировали значительно большую дисперсию (рис.2 C и фильмы S3 и S4).
Рис. 2.Связанные с кластером PMNs обнаруживают миграционное ограничение, которое зависит от аутологичного хемотаксиса к секретируемым факторам. ( A ) Связанные с кластером и свободные PMN арестовываются внутримышечно. ( B ) Скорость миграции и сквозное расстояние от исходного положения связанных с кластером и свободных PMN ( n = 43–53 PMN на условие для пяти устройств). ( C ) Дорожки свободных (синий) и связанных с кластером (красный) PMN за 90 минут (временной шаг 40 с).Пунктирным кружком обозначен радиус 150 мкм. ( D ) Набор цитокинов, показывающий относительные величины секретируемых факторов только из MA2, только PMN, LPS-активированных PMN или совпадения MA2 + PMN после 4 ч культивирования (два повтора). ( E ) Скорость миграции и сквозное расстояние от исходного положения связанных с кластером PMN с или без анти-CXCL-1 + анти-IL-8 ( n = 30–34 PMN на условие, более пяти устройств). Планки погрешностей указывают на SD, ** P <0.01, *** P <0,001. ( F ) Треки миграции кластер-ассоциированных PMN, инкубированных с анти-CXCL-1 + анти-IL-8 или контрольным (Ctrl) IgG.
Наборы цитокинов выявили два высоко выраженных хемотаксических фактора нейтрофилов, IL-8 (из MA2 и воспаленных PMN) и CXCL-1 (из MA2) (рис. 2 D и SI, приложение , рис. S6 A ) , а при нейтрализации антителами в устройствах мы наблюдали увеличение дисперсии, скорости миграции и смещения PMN (рис.2 E и F ), а также увеличенное расстояние разделения от периферии кластера TC – PMN ( SI Приложение , рис. S5 A и B ). Характер дисперсии PMN сохранялся до 6 часов (рис. 3 A ). Таким образом, вероятно, что кластерные клетки могут вести себя как концентрированный источник хемокинов для близлежащих PMN. Затем мы расшифровали относительные уровни вкладов отдельных хемокинов и их источников. В то время как блокирование одного CXCL-1 приводило к значительному снижению дисперсии, блокирование одного IL-8 приводило к дисперсии на уровне, аналогичном коблокированию (рис.3 В ). Это говорит о том, что IL-8 является более сильным хемокином, чем CXCL-1 в условиях заключения; однако потеря IL-8 может быть немного компенсирована активностью CXCL-1 или других хемокинов. Поскольку IL-8 секретируется как стимулированными PMN, так и MA2 на высоких уровнях ( SI, приложение , рис. S8, B и C ), мы разделили два источника через siRNA. Интересно, что удаление IL-8, происходящего из опухоли, существенно не повлияло на секвестрацию нейтрофилов (фиг. 3 C ).Замена TC гранулами размером 15 мкм не привела к изменению дисперсии, тогда как обработка гранул и PMN анти-IL-8 вызвала высокие уровни дисперсии, что позволяет предположить, что IL-8, полученный из PMN, достаточен для индукции хемотаксического ограничения. Моделирование COMSOL также подтвердило, что градиенты IL-8 могут поддерживаться в присутствии потока ( SI Приложение , рис. S9). Более того, IL-8, секретируемый активированными PMN, может быть изолирован внутри гликокаликса, покрывающего поверхность эндотелия.Это может помочь поддерживать достаточный уровень IL-8 в потоке, о чем свидетельствует значительная дисперсия PMN из кластеров (без изменений скорости миграции) при предварительной обработке сосудов гепариназой III (рис.3 C и SI, приложение , рис. S10).
Рис. 3.Ограниченная миграция PMN секвестрирует PMN вблизи TC в течение> 6 часов и усиливается эндотелиальным гликокаликсом. ( A ) Связанные с кластером PMN и их дисперсия из TC в течение 2 часов в отсутствие и в присутствии анти-CXCL-1 + анти-IL-8.( B ) Фракция PMN, остающихся в отдельных кластерах в течение 3 часов (каждые 10 минут), когда система обрабатывается нейтрализующими антителами IL-8 и / или CXCL-1, или когда опухолевые клетки заменяются полистирольными гранулами 15 мкм ( n = 15 кластеров на группу на трех независимых устройствах в каждый момент времени). ( C ) Доля PMN, остающаяся на кластер TC – PMN через 6 часов. Устройства обрабатываются либо IgG, либо анти-IL-8 и / или анти-CXCL-1. В некоторых случаях только опухолевый IL-8 и / или CXCL-1 удаляется через миРНК.Каждая точка представляет одно устройство с по крайней мере пятью кластерами TC – PMN, усредненными на одно устройство ( n > 5 устройств на одно условие). ** P <0,01, *** P <0,001, н.у. = незначительно, ошибка = SEM.
Трансмиграционные внесосудистые PMN (которые в дальнейшем были разделены на свободные популяции и популяции, связанные с кластерами) демонстрировали характер дисперсии, аналогичный соответствующим случаям, когда внутри- и внесосудистые PMN не различались. Кроме того, свободные PMN имели гораздо более высокую вероятность экстравазации по сравнению с PMN, связанными с кластером, в то время как блокирование IL-8 и CXCL-1 увеличивало частоту экстравазации связанных с кластером PMN ( SI Приложение , рис.S11 и фильм S5). В совокупности это предполагает, что различия в доступности рецепторов адгезии на эндотелии (например, ICAM-1) не влияют на удержание PMN.
Таким образом, после агрегации TC – PMN посредством механизмов улавливания и адгезии PMN далее секвестрируются с помощью хемотаксического ограничения, опосредованного самовоспроизводящимся IL-8 и происходящим из опухоли CXCL-1. Иммобилизация хемокинов в эндотелиальном гликокаликсе может дополнительно способствовать увеличению доступности IL-8 и, следовательно, ограничению миграции.
Пространственная локализация нейтрофилов и секретируемых ими факторов способствует локальной экстравазации опухолевых клеток.
Меланомы A375 и A375-MA2 и MDA-MB-231 молочной железы коперфузировали либо с покоящимися, либо с LPS-стимулированными PMN. В то время как системная инъекция ЛПС вызывала самый высокий уровень экстравазации ( SI Приложение , рис. S6 D ), коперфузия только с предварительно стимулированными ЛПС PMN также значительно увеличивала экстравазацию во всех трех клеточных линиях, что указывает на прямой эффект только от стимулированных PMN. (Рисунок.4 А ). Важно отметить, что частота экстравазации PMN-ассоциированных клеток была выше по сравнению с PMN-свободными клетками, что позволяет предположить наличие зависимого от близости эффекта PMN на соседние TC и / или EC (рис. 4 B ). Нейтрализация IL-8 не только устраняет секвестр PMN, но также снижает частоту экстравазации TCs, связанных с PMN. Отсутствует влияние на частоту экстравазации свободной субпопуляции, что указывает на прямую корреляцию между IL-8-зависимым ограничением миграции и экстравазацией.Инкубация устройства с кондиционированной средой только от стимулированных PMN или сокультуры MA2-PMN вызвала аналогичное увеличение экстравазации (рис. 4 C ). Таким образом, миграционное ограничение PMN может облегчить пространственную локализацию производных PMN факторов проэкстравазации в близлежащих арестованных TC и повысить их трансмиграционный потенциал.
Рис. 4.IL-8, высвобождаемый LPS-стимулированными PMN, увеличивает частоту экстравазации TC за счет действия IL-8 на локальную барьерную функцию EC. ( A ) Процент экстравазированных клеток через 6 часов для A375, A375-MA2 и MDA-MB-231 при коперфузии с покоящимися PMN или LPS-стимулированными PMN в соотношении 1: 5 ( n = 12 устройств).( B ) Процент экстравазированного MA2 в PMN-ассоциированных или неассоциированных субпопуляциях в одном и том же устройстве, с или без анти-IL-8 ( n = 6 устройств). ( C ) Скорость экстравазации MA2 через 6 часов при совместном использовании с кондиционированной средой (CM), полученной в различных условиях ( n > 9 устройств на условие). ( D ) Скорость экстравазации MA2 в присутствии стимулированных PMN и нейтрализующих антител через 6 часов после инъекции ( n = 5 устройств).( E ) Проницаемость микрососудов для декстрана 70 кДа после 6-часовой обработки стимулированной кондиционированной средой PMN. Скорость экстравазации (через 6 часов) опухолевых клеток MA2 в предварительно обработанных PMN средах (в течение 6 часов) ( n = 3 устройства). ( F ) Процент исходных (при т = 0) PMN, остающихся в слоях микрососудов после 6 часов потока в отсутствие или в присутствии анти-IL-8 + анти-CXCL-1 ( n = 10 устройств ). ( G ) Процент PMN, оставшихся в связанных с кластером или свободных субпопуляциях после 6 часов кровотока (с коперфузией MA2).Каждая точка представляет собой среднее значение по крайней мере пяти кластеров по пяти интересующим регионам (ROI) или по крайней мере 20 бесплатных PMN по пяти ROI. В A — G планки ошибок указывают SEM, * P <0,05, ** P <0,01, *** P <0,001.
Коперфузия нестимулированных PMN также привела к увеличению экстравазации (хотя и в меньшей степени) у A375 и MA2, но не у MDA-MB-231. Поскольку экспрессия ICAM-1 была связана с повышенной злокачественностью некоторых меланом (27), возможно, что опухоль ICAM-1 могла автономно модулировать экстравазацию, а более высокая экспрессия ICAM-1 в определенных линиях опухолевых клеток могла снизить потребность в PMN и системное воспаление экстравазируется по сравнению с опухолями с более низким уровнем.Мы проверили частоту экстравазации панели клеточных линий, включая меланомы (A375, A375-MA2 и WM266-4), толстой кишки (HT29 и SW620) и немелкоклеточный рак легкого (NSCLC) (h3126 и h3087), а также их корреляцию с Экспрессия на поверхности ICAM-1, измеренная с помощью проточной цитометрии. В целом, потенциалы экстравазации положительно коррелируют с уровнями ICAM-1 для тестируемых линий меланомы и NSCLC, но не для линий толстой кишки, что указывает на специфичность опухолевого типа ( SI Приложение , Рис. S6 B ).Нейтрализация опухоли ICAM-1 приводит к уменьшению экстравазации MA2, но не A375 или MDA-MB-231, предполагая, что опухоль ICAM-1 может модулировать экстравазацию независимо от инфекции, но, возможно, только тогда, когда ICAM-1 экспрессируется на достаточном уровне. Более того, в то время как опухоль ICAM-1 действительно увеличивает уровни агрегации и остановки опухоли с PMN в капиллярах, взаимодействие ICAM-1 с PMN может стимулировать эффекты экстравазации протопухолевых клеток в PMN в отсутствие системной инфекции. Когда меланома ICAM-1 нейтрализуется в присутствии покоящихся PMN, частота экстравазации снижается, но не достигает уровней, наблюдаемых при блокировании TC ICAM-1 в отсутствие PMN.Таким образом, PMN могут оказывать, по крайней мере частично, эффект проэкстравазации, который не происходит из-за наличия опухоли ICAM-1. Наконец, блокирование ICAM-1 в присутствии MA2 и LPS-стимулированных PMN не ослабляет экстравазацию, указывая на то, что вызванное инфекцией увеличение экстравазации не зависит от ICAM-1 ( SI Приложение , рис. S6 C — E ). Таким образом, остается ясно, что независимо от роли опухоли ICAM-1, системное воспаление резко увеличивает экстравазацию, и ICAM-1, по-видимому, не играет критической роли в этом случае.
ИЛ-8, производный от PMN, играет множественную роль в секвестрации PMN и усилении экстравазации опухолевых клеток.
Из цитокинов, активируемых при стимуляции PMN с помощью стимуляции LPS или совпадения с MA2 (IL-8, IL-6, CCL4 и CXCL-7), только блокирование IL-8 снижает трансмиграцию (рис. 4 D). ). Обработка устройством PMN-кондиционированной средой увеличивала экстравазацию, в то время как совместная обработка кондиционированной среды с анти-IL-8 ослабляла этот ответ, указывая на то, что IL-8, производный от PMN, ответственен за усиление экстравазации контактно-независимым образом (рис.4 E ). Это также коррелирует с увеличением проницаемости сосудов, когда сосуды предварительно обрабатываются PMN-кондиционированной средой, которая ослабляется анти-IL-8 (рис. 4 E ).
Поскольку MA2 продуцирует IL-8 в той же степени, что и стимулированные PMN, мы спросили, могут ли PMN, полученные исключительно из IL-8, быть достаточными для усиления экстравазации, независимо от уровней IL-8, полученных из опухоли. Коперфузия МА2, обработанных миРНК IL-8, и ЛПС-стимулированных ПМЯ не уменьшала частоту экстравазации ( SI Приложение , рис.S8), что указывает на то, что уровень одного IL-8, производного от MA2, может быть ниже порогового уровня, чтобы вызвать заметные последствия при трансмиграции. Таким образом, помимо модуляции хемотаксического ограничения, производный от PMN IL-8 может играть дополнительную роль в облегчении экстравазации через нарушение функции эндотелиального барьера.
Наконец, повышенная дисперсия PMN из кластеров коррелирует с потерей PMN из всего микрососудистого русла (рис. 4 F ). Потеря свободных PMN была значительно выше по сравнению с PMN, которые изначально были связаны с кластером, что позволяет предположить, что секвестрированные PMN более защищены от сдвигового потока (рис.4 G ). Таким образом, миграционное ограничение PMN в кластерах может увеличить количество PMN, задержанных в микрососуде, и максимизировать проэкстравазационный эффект PMN.
Воспаленные PMN демонстрируют ограниченную миграцию и усиливают экстравазацию опухолевых клеток в эмбрионах рыбок данио.
Подобно модели сосуда на чипе, когда LPS-стимулированные нейтрофилы совместно инъецируются с A375 или MA2 в эмбрионы рыбок данио, PMNs быстро задерживаются в гетеротипных агрегатах, и большинство из них остаются в покое в течение периодов> 6 часов ( SI Приложение , рис. .5 A и Movie S6). Примечательно, что мы наблюдали аналогичное поведение удержания связанных с кластером PMN по сравнению со свободными PMN (рис. 5 B ). В отличие от модели на чипе, нейтрофилы продолжают циркулировать по всему эмбриону с течением времени. Несмотря на это, мы наблюдали, что очень мало нейтрофилов рекрутируется в существующие кластеры с течением времени, поскольку закупорка сосуда, по-видимому, предотвращает дальнейший значительный приток крови в окрестности. Наконец, мы подтвердили, что совместная инъекция MA2 и A375 со стимулированными человеческими PMN увеличивала скорость экстравазации опухолевых клеток в течение 12 часов, аналогично нашим результатам, полученным in vitro (рис.5 С ).
Рис. 5.Воспаленные PMN секвестрируются в кластерах TC – PMN и увеличивают скорость экстравазации A375 и MA2 in vivo. ( A ) Связанные с внутрисосудистым кластером и свободные PMN (розовый) при совместной инъекции с клетками MA2 (зеленый) в flk: dsRed (красный) эмбрионах рыбок данио. ( B ) Следы миграции ассоциированных с кластером или свободных PMN в сосудах эмбрионов рыбок данио. ( C ) Частота экстравазации клеток A375 и MA2 с коперфузией или без нее с воспаленными PMN через 6 и 24 часа ( n = 7–12 эмбрионов для A375, n = 18–23 эмбриона для MA2).Планки погрешностей указывают на SEM, ** P <0,01, *** P <0,001.
Обсуждение
Хотя роль нейтрофилов в очаге первичной опухоли широко изучена, вопрос о том, играют ли нейтрофилы про- или антиметастатические функции во время гематогенного распространения, менее ясно. Воспалительная активация в ходе прогрессирования опухоли может возникнуть в результате инфекций в результате лечебных вмешательств, таких как операции резекции (18), стерильного воспаления в результате химиотерапии (19) и даже кондиционирования самой первичной опухолью (3).Важно отметить, что воспаление часто коррелирует с неблагоприятными онкологическими исходами независимо от заболеваемости, связанной с воспалительной реакцией (12–14). Таким образом, нейтрофилы могут играть важную роль в связывании инфекции, воспаления и метастазирования. В этом исследовании мы стремились понять динамическую роль воспаленных нейтрофилов во время остановки опухолевых клеток и экстравазации.
Мы используем модель микроциркуляторного русла человека in vitro, что позволяет нам динамически визуализировать взаимодействия опухоль-PMN-эндотелий с высоким пространственно-временным разрешением, что в противном случае было бы затруднительно in vivo.Мы можем резюмировать ключевые события, такие как остановка опухолевых клеток и нейтрофилов в сосудах, диаметр и характер потока которых сравним с таковыми в сосудистой сети in vivo (23, 24). Это ключевой момент, поскольку кластеризация TC – PMN, секвестрация PMN и образование градиентов хемокинов не могут быть воспроизведены в статических анализах или анализах, связанных с потоком через плоский эндотелиальный слой. Кроме того, тонкая высота наших тканей (<150 мкм) облегчает отслеживание быстро мигрирующих нейтрофилов по сравнению с визуализацией толстых тканей in vivo.Это позволяет нам определять пространственные отношения между отдельными клетками и разрешать различное миграционное поведение между клеточными субпопуляциями, такое как аутологичный хемотаксис нейтрофилов в захваченных кластерах.
Ранее на мышиных моделях диссеминации было показано, что задержанные опухолевые клетки могут колокализоваться с нейтрофилами посредством интегринов (8, 9, 28, 29), NET (18) или притяжения тромбоцитов (7). Донг с соавторами (8) показали, что опухолевые клетки ICAM-1 и PMN CD11b / beta-2 способствовали агрегации TC-PMN в камере с параллельным потоком.Однако на самом деле TC и PMN взаимодействуют в узких капиллярах (а не на плоской поверхности), где могут играть роль физическая окклюзия и увеличенная площадь контакта с эндотелием. Объединив оба механизма в нашем анализе, мы также обнаружили, что повышающая регуляция эндотелиального ICAM-1 при контакте с TC и PMN может увеличивать скорость ассоциации и дает более сильный эффект, чем опухолевый ICAM-1. Важно, что физическая окклюзия может играть значительную роль в кластеризации TC – PMN, поскольку замена PMN на 4-мкм бусинки в микрососудах по-прежнему приводит к нетривиальным уровням ассоциации TC – бусинок.
Начальная кластеризация TC – PMN в потоке может быть важной предпосылкой для хемотаксической секвестрации PMN. Вместо того, чтобы формировать стабильные адгезии к TC после задержания, связанные с кластером нейтрофилы мигрируют «назад и вперед» в радиусе ~ 70 мкм от исходного кластера TC-PMN посредством механизма ограничения миграции, который зависит от IL, полученного из PMN. -8. Интересно, что это поведение существует даже без заметного рекрутирования нейтрофилов из других частей сосудистого русла, поскольку начальные уровни PMN в кластерах, образованных во время внутрисосудистой остановки, вероятно, достаточны для установления источника хемокинов.
Ограничение PMN в заблокированных опухолевых кластерах может иметь ключевое значение для усиления проэкстравазационных эффектов PMN. Воспаленные PMN секретируют высокий уровень IL-8, который, помимо ограничения миграции, может еще больше разрушить эндотелиальный барьер, облегчая трансмиграцию опухолевых клеток. Кроме того, хотя МА2 также секретирует ИЛ-8, похоже, что большая часть проэкстравазационного эффекта ИЛ-8 возникает только от PMN. Субпопуляция ОК, ассоциированная с PMN, демонстрирует заметно более высокую частоту экстравазации, что свидетельствует о зависимом от близости влиянии PMN на экстравазацию.Вероятно, что секвестрация PMN в остановленных опухолевых очагах позволяет IL-8 быть высоко локализованным и эффективно воздействовать на соседний эндотелий. Значительное увеличение количества колокализованных PMN на опухолевый кластер после лечения LPS может еще больше усугубить этот эффект. Кроме того, частота экстравазации в PMN-ассоциированных TC снижалась после нейтрализации хемотаксических факторов IL-8 и CXCL-1, но не в неассоциированной популяции, что указывает на то, что ограничение PMN имеет решающее значение для проявления эффектов IL-8, производного от PMN.Фактически, меланома IL-8 участвует в разложении VE-кадгерина (30). Хотя мы также обнаружили, что IL-8 является ключевым эффектором в нашей системе, наше исследование затрагивает IL-8, производный от PMN (а не от TC), и прямой эффект его секвестрации около опухолевых клеток на облегчение трансэндотелиальной миграции опухолевых клеток.
Мы используем LPS-стимулированные нейтрофилы для имитации воспаленного фенотипа, который обычно обнаруживается при воспалении, вызванном инфекцией. Однако воспаленное состояние нейтрофилов может быть неоднородным в зависимости от источника воспалительного стимула (например,g., стерильное воспаление, обусловленное первичной опухолью). Интересно, что даже без предварительной стимуляции PMN все еще способны повышать скорость экстравазации опухолевых клеток (хотя и в меньшей степени, чем стимулированные PMN). Таким образом, опухолевые клетки могут обладать способностью активировать PMN с образованием факторов проэкстравазации (8), которые не зависят от инфекции. Это очень вероятно в случаях стерильного воспаления или когда нейтрофилы обусловлены первичными опухолями. Чтобы поддержать это, PMN, предварительно стимулированные кондиционированной опухолью средой, также усиливают экстравазацию.Тем не менее, PMN, активированные инфекцией, могут способствовать экстравазации в гораздо большей степени, чем эффекты одних только опухолевых клеток, и, по-видимому, делают это без специфичности опухолевых клеток.
Используя модель микроциркуляторного русла человека in vitro, мы показали, что воспаленные PMN и опухолевые клетки могут задерживаться в гетеротипических кластерах из-за механического захвата в узких капиллярах в сочетании с образованием адгезии между PMN и эндотелиальным ICAM-1, активируемым при перфузии опухолевых клеток. Связанные с кластером PMN не образуют постоянных контактов с TC или EC, а скорее мигрируют ограниченным образом, который опосредован аутологичным хемотаксисом к самовоспроизводящемуся IL-8 и усиливается присутствием гликокаликса EC.Следовательно, PMN секвестрируются, что приводит к пространственной локализации производного от PMN IL-8, который играет дополнительную роль в разрушении эндотелиального барьера и повышении потенциала экстравазации близлежащих арестованных опухолевых клеток ( SI, приложение , рис. S13).
Материалы и методы
Разработка и изготовление устройств.
Используемый микрожидкостный чип представляет собой версию с более высокой пропускной способностью нашей предыдущей конструкции (23), которая служит для повышения пропускной способности, надежности и согласованности формирования микрососудистой сети.Один порт для инъекции геля позволяет параллельное распределение клеток по восьми независимым сосудистым ложам через разветвленную сеть (Рис. 1 A и SI Приложение ).
Анализ экстравазации эмбрионов рыбок данио.
Анестезированным эмбрионам (линия flk: dsRed2 , пересеченная с фоном casper ) инъецировали 4 нл 4 × 10 4 опухолевых клеток на микролитр в PBS или 4 × 10 4 опухолевых клеток и 2 × 10 5 человеческих предварительно стимулированных ЛПС PMN на микролитр в PBS.Эмбрионы помещали в одну лунку шестилуночного планшета со стеклянным дном (MatTek), содержащего тонкий слой 2% агарозы, и покрывали 0,8% агарозой, содержащей 0,01% трикаина, забуференного до pH 7,4. Эмбрионы получали на инвертированном конфокальном микроскопе A1R (Nikon) с использованием резонансного сканера с 10-кратным объективом и 1,5-кратным увеличением.
Выделение нейтрофилов человека.
Человеческие нейтрофилы были получены из свежей человеческой крови (разведенной 1: 1 в HBSS; Invitrogen), приобретенной у Research Blood Components, которая была взята в соответствии с рекомендациями Американской ассоциации банков крови.Субъекты состоят из здоровых мужчин и женщин в возрасте от 18 до 65 лет. От каждого донора была получена форма согласия, одобренная институциональным наблюдательным советом, дающая разрешение на использование их образцов в исследовательских целях. PMN выделяли с использованием традиционного градиента Histopaque 1077 (Sigma).
Статистика.
Статистический анализ проводился с помощью SigmaPlot с использованием двустороннего критерия Стьюдента t при сравнении двух условий или однофакторного дисперсионного анализа с апостериорным анализом Тьюки, когда это применимо.
Сноски
Вклад авторов: M.B.C., D.C.B., R.O.H. и R.D.K. спланированное исследование; M.B.C., C.H., D.C.B. и C.Y. проведенное исследование; Х.А. внесены новые реагенты / аналитические инструменты; M.B.C., C.H. и C.Y. проанализированные данные; и M.B.C., C.H., D.C.B. и R.D.K. написал газету.
Авторы заявляют об отсутствии конфликта интересов.
Эта статья представляет собой прямое представление PNAS.
Эта статья содержит вспомогательную информацию на сайте www.pnas.org/lookup/suppl/doi:10.1073/pnas.1715932115/-/DCSupplemental.
Mac-1 (CD11b / CD18) в миграции нейтрофилов — Проекты — Kim Lab
URMC / Labs / Kim Lab / Projects / Mac-1 (CD11b / CD18) в миграции нейтрофилов
Нейтрофилы играют ключевую роль в системе защиты хозяина от патогенов в силу их способности фагоцитировать микроорганизмы и продуцировать свободные радикалы кислорода и протеолитические ферменты.Экстравазация нейтрофилов из кровотока проходит через три скоординированных этапа: перекатывание и связывание, прочная адгезия и трансмиграция. Первый шаг зависит от молекул селектина, экспрессируемых как на нейтрофилах, так и на эндотелиальных клетках (ЭК). Второй этап опосредуется посредством взаимодействия интегринов β 2 , включая LFA-1 и Mac-1, с их контррецепторами, ICAM-1 и ICAM-2, на EC. Затем за процессом следует дополнительный этап, называемый «трансэндотелиальной миграцией», при котором нейтрофилы покидают кровоток и попадают в место воспаления.Важность семейства 2 интегринов in vivo иллюстрируется пациентами с наследственной недостаточностью адгезии лейкоцитов (LAD), у которых имеется генетический дефект в экспрессии интегрина 2 . Пациенты с LAD страдают рецидивирующими, опасными для жизни бактериальными инфекциями и нарушением заживления ран и являются кандидатами на трансплантацию костного мозга в раннем возрасте.
Изображения CFP и YFP клеток венулы Cremaster живой мыши.
Недавние исследования переноса лейкоцитов позволили существенно понять физиологические и патологические паттерны миграции лейкоцитов и их роль в здоровье и болезнях.Эта информация была важна для разработки новых противовоспалительных методов лечения, таких как моноклональные антитела против интегринов лейкоцитов для лечения псориаза и рассеянного склероза. Среди установленных методов изучения движения лейкоцитов прямое наблюдение за прокаткой и миграцией лейкоцитов с помощью прижизненной микроскопии (IVM) является одним из наиболее важных экспериментальных подходов. Один из методов изучения различных подгрупп лейкоцитов состоит в очистке клеток от мышей-доноров, их флуоресцентной метке in vitro и повторной инъекции реципиенту.Трансгенные мыши, экспрессирующие зеленый флуоресцентный белок (GFP) или его варианты в зависимости от типа клеток, являются альтернативой очищенным ex vivo флуоресцентно меченным клеткам.
Этот подход подвергает клетки воздействию менее ex vivo , и генетически кодируемые флуоресцентные метки не растворяются и не исчезают в делящихся клетках. Кроме того, флуоресцентные белковые трансгенные лейкоциты можно изучать эндогенно без манипуляций ex vivo или адоптивного переноса.Флуоресцентные сенсоры in vivo, основанные на FRET, обещают, что IVM будет изучать активацию клеток и / или передачу сигналов во время длительных миграций клеток. В настоящее время мы создаем множество мышей с нокаутом флуоресцентного белка для изучения динамической миграции нейтрофилов и активации интегрина у живых животных.
11.3G: Воспаление — Биология LibreTexts
Воспалительная реакция — это попытка организма восстановить и поддерживать гомеостаз после травмы и является неотъемлемой частью защиты организма.Большинство защитных элементов организма находится в крови, и воспаление — это средство, с помощью которого защитные клетки организма и защитные химические вещества покидают кровь и попадают в ткани вокруг травмированного или инфицированного участка. Воспаление существенно полезно, однако чрезмерное или продолжительное воспаление может нанести вред.
Механизм воспаления
По сути, четыре процесса составляют воспалительный механизм:
а. Гладкие мышцы вокруг более крупных кровеносных сосудов сокращаются, чтобы замедлить кровоток через капиллярные русла в инфицированном или поврежденном месте.Это дает лейкоцитам больше возможностей прилипнуть к стенкам капилляра и выдавиться в окружающие ткани.
г. Эндотелиальные клетки, составляющие стенки более мелких кровеносных сосудов, сокращаются. Это увеличивает пространство между эндотелиальными клетками, что приводит к увеличению проницаемости капилляров. Поскольку в результате этого кровеносные сосуды становятся больше в диаметре, этот процесс называется вазодилатацией (см. Рисунок \ (\ PageIndex {1} \)).
Микрофотографии с помощью сканирующего электронного микроскопа поперечного сечения капилляра, показывающие эндотелиальную клетку и капилляр с эритроцитом; любезно предоставлено микроскопией Денниса Кункеля).
г. Молекулы, называемые селектинами, продуцируются на мембране лейкоцитов и способны обратимо связываться с соответствующими рецепторами гликопротеинов селектина на внутренней стенке венулы. Это обратимое связывание позволяет лейкоциту катиться по внутренней стенке венулы. Это обратимое связывание позволяет лейкоциту катиться по внутренней стенке венулы. Молекулы адгезии активируются на поверхности эндотелиальных клеток на внутренней стенке капилляров. Соответствующие молекулы на поверхности лейкоцитов, называемые интегринами, прикрепляются к этим молекулам адгезии, позволяя лейкоцитам уплощаться и продавливаться через пространство между эндотелиальными клетками.Этот процесс называется диапедезом или экстравазией.
г. Активация пути коагуляции заставляет фибриновые сгустки физически улавливать инфекционные микробы и предотвращать их попадание в кровоток. Это также вызывает свертывание крови в окружающих мелких кровеносных сосудах, чтобы остановить кровотечение и дополнительно предотвратить попадание микроорганизмов в кровоток.
Видеоролик YouTube Tube и анимация экстравазации лейкоцитов (диапедез) |
3D-анимация, иллюстрирующая выход лейкоцитов из капилляров и вход в ткань (а также эндопротез) в лейкоците. |
Эти четыре события запускаются и усиливаются различными химическими медиаторами воспаления. Теперь мы разделим воспалительную реакцию на две стадии: раннее воспаление и позднее воспаление.
Раннее воспаление и диапедез
В большинстве случаев диапедез лейкоцитов (экстравазация) происходит в посткапиллярных венулах, потому что в этих венулах ниже гемодинамические силы сдвига.Это облегчает прикрепление лейкоцитов к внутренней стенке сосуда и выдавливание между эндотелиальными клетками. Мы рассмотрим этот процесс более подробно ниже.
- На самых ранних стадиях воспаления стимулы, такие как травма или инфекция, вызывают высвобождение различных медиаторов воспаления, таких как лейкотриены, простагландины и гистамин. Связывание этих медиаторов с их рецепторами на эндотелиальных клетках приводит к расширению сосудов, сокращению эндотелиальных клеток и увеличению проницаемости кровеносных сосудов.Кроме того, базальная мембрана, окружающая капилляры, перестраивается, чтобы способствовать миграции лейкоцитов и перемещению макромолекул плазмы из капилляров в окружающую ткань. Тучные клетки соединительной ткани, а также базофилы, нейтрофилы и тромбоциты, покидающие кровь из поврежденных капилляров, высвобождают или стимулируют синтез сосудорасширяющих средств, таких как гистамин, лейкотриены, кинины и простагландины. Определенные продукты путей комплемента (C5a и C3a) могут связываться с тучными клетками и запускать высвобождение их вазоактивных агентов.Кроме того, повреждение тканей активирует каскад коагуляции и выработку медиаторов воспаления, таких как брадикинины.
- Связывание гистамина с рецепторами гистамина на эндотелиальных клетках запускает повышающую регуляцию молекул Р-селектина и фактора активации тромбоцитов или PAF на эндотелиальных клетках, выстилающих венулы.
- Р-селектины затем способны обратимо связываться с соответствующими лигандами гликопротеина Р-селектина (PSGL-1) на лейкоцитах. Это обратимое связывание позволяет лейкоциту катиться по внутренней стенке венулы.
- Связывание PAF с его соответствующим рецептором PAF-R на лейкоците усиливает поверхностную экспрессию интегрина, называемого молекулой-1, ассоциированной с функцией лейкоцитов (LFA-1), на поверхности лейкоцита.
- Молекулы LFA-1 на катящихся лейкоцитах теперь могут прочно связываться с молекулой адгезии, называемой молекулой межклеточной адгезии-1 (ICAM-1), обнаруженной на поверхности эндотелиальных клеток, образующих внутреннюю стенку кровеносного сосуда (см. Рисунок \ (\ PageIndex {4} \)).
- Лейкоциты выравниваются, протискиваются между суженными эндотелиальными клетками и используют ферменты для разрушения матрикса, образующего базальную мембрану, окружающую кровеносный сосуд. Затем лейкоциты перемещаются к хемотаксическим агентам, таким как белок комплемента C5a и лейкотриен B 4 , генерируемый клетками в месте инфекции или повреждения (см. Рисунок \ (\ PageIndex {5} \)).
Позднее воспаление и диапедез
1) Обычно в течение двух-четырех часов после ранних стадий воспаления активированные макрофаги и эндотелиальные клетки сосудов высвобождают воспалительные цитокины, такие как TNF и IL-1, когда их толл-подобные рецепторы связывают молекулярные патогены. паттерны — молекулярные компоненты, связанные с микроорганизмами, но не входящие в состав эукариотических клеток.Это позволяет эндотелиальным клеткам соседних венул увеличивать экспрессию молекул адгезии, таких как Р-селектины, Е-селектины, молекулы межклеточной адгезии (ICAM) и хемокины.
2) Связывание TNF и IL-1 с рецепторами на эндотелиальных клетках запускает и поддерживает воспалительный ответ за счет усиления выработки молекулы адгезии E-селектина и поддержания экспрессии P-селектина на эндотелиальных клетках, выстилающих венулы.
3). E-селектины на внутренней поверхности эндотелиальных клеток теперь могут прочно связываться с соответствующим интегрином E-селектиновым лигандом-1 (ESL-1) на лейкоцитах (см. Рисунок \ (\ PageIndex {4} \)).
4) Лейкоциты выравниваются, сжимаются между суженными эндотелиальными клетками и перемещаются через базальную мембрану, поскольку их привлекают хемокины, такие как интерлейкин-8 (IL-8) и хемотаксический белок моноцитов-1 (MCP-1). генерируется клетками в месте заражения или травмы (см. Рисунок \ (\ PageIndex {5} \)). Утечка фибриногена и фибронектина плазмы затем формирует молекулярный каркас, который усиливает миграцию и удержание лейкоцитов на инфицированном участке.
Преимущества воспаления
В результате повышенная проницаемость:
а.Плазма выходит из крови в ткани.
Полезные молекулы в плазме (см. Рисунок \ (\ PageIndex {2} \)) включают:
1. Факторы свертывания. Повреждение ткани активирует каскад коагуляции, вызывая образование фибриновых сгустков для локализации инфекции, остановки кровотечения и хемотаксического привлечения фагоцитов.
2. Антитела. Они помогают удалить или заблокировать действие микробов с помощью различных методов, которые будут объяснены в Разделе 6.
3. Белки путей комплемента.Они, в свою очередь: 1) стимулируют большее воспаление (C5a, C3a и C4a), 2) прикрепляют микроорганизмы к фагоцитам (C3b и C4b), 3) хемотаксически привлекают фагоциты (C5a) и 4) лизируют мембраносвязанные клетки, демонстрирующие чужеродные антигены (комплекс мембранной атаки или МАК).
4. Питательные вещества. Они питают клетки воспаленной ткани.
5. Лизоцим, кателицидины, фосфолипаза А 2 и дефенсины человека. Лизоцим разрушает пептидогликан. Кателицидины расщепляются на два пептида, которые непосредственно токсичны для микробов и могут нейтрализовать ЛПС из грамотрицательной бактериальной клеточной стенки.Фосфолипаза A 2 гидролизует фосфолипиды в цитоплазматической мембране бактерий. Дефенсины человека создают поры в цитоплазматических мембранах многих бактерий. Дефенсины также активируют клетки, участвующие в воспалительной реакции.
6. Трансферрин. Трансферрин лишает микробы необходимого железа.
г. Лейкоциты попадают в ткань посредством процесса, называемого диапедезом или экстравазацией, который обсуждался выше в разделе «Раннее воспаление» и «Позднее воспаление».
Преимущества диапедеза включают (см. Рисунок \ (\ PageIndex {2} \)):
1.Повышенный фагоцитоз. Нейтрофилы, моноциты, которые при попадании в ткань дифференцируются в макрофаги, и эозинофилы являются фагоцитарными лейкоцитами.
2. Расширение сосудов. Базофилы, эозинофилы, нейтрофилы и тромбоциты проникают в ткань и высвобождают или стимулируют выработку вазоактивных агентов, способствующих воспалению.
3. Цитотоксические Т-лимфоциты (CTL), эффекторные Т4-клетки и NK-клетки проникают в ткань, чтобы убить клетки, такие как инфицированные клетки и раковые клетки, которые отображают чужеродные антигены на своей поверхности (обсуждается в Блоке 6).
Цитокины, называемые хемокинами, особенно важны в этой части воспалительной реакции. Они играют ключевую роль в диапедезе, позволяя лейкоцитам прилипать к внутренней поверхности кровеносных сосудов, мигрировать из кровеносных сосудов в ткань и хемотаксически привлекаться к поврежденному или инфицированному месту. Они также вызывают нейтрофилы внеклеточного уничтожения.
Наконец, в течение 1-3 дней макрофаги высвобождают цитокины интерлейкин-1 (IL-1) и фактор некроза опухоли альфа (TNF-a).Эти цитокины стимулируют NK-клетки и Т-лимфоциты, чтобы производить цитокин интерферон-гамма. (ЕСЛИ-?). ЕСЛИ-? затем связывается с рецепторами на макрофагах, заставляя их продуцировать фактор роста фибробластов и ангиогенные факторы для ремоделирования ткани. По мере разрастания эндотелиальных клеток и фибробластов эндотелиальные клетки образуют тонкую сеть новых капилляров в поврежденной области, чтобы снабжать воспаленную ткань кровью, кислородом и питательными веществами. Фибробласты откладывают белковый коллаген в поврежденной области и образуют мост из соединительной рубцовой ткани, закрывающий открытую открытую область.Это называется фиброзом или рубцеванием и представляет собой заключительную стадию заживления.
Воспаление обычно тщательно регулируется цитокинами. Воспалительные цитокины, такие как гамма-интерферон и интерлейкин-12, усиливают воспалительную реакцию, тогда как цитокин интерлейкин-10 подавляет воспаление, снижая экспрессию воспалительных цитокинов.
Итак, как можно видеть, острое воспаление необходимо для защиты организма. Однако хроническое воспаление может привести к значительному повреждению тканей и рубцеванию.При длительном увеличении проницаемости капилляров нейтрофилы постоянно покидают кровь и накапливаются в ткани на инфицированном или поврежденном участке. Когда они высвобождают лизосомное содержимое и активные формы кислорода или АФК, окружающие ткани разрушаются и в конечном итоге заменяются рубцовой тканью. Противовоспалительные агенты, такие как антигистаминные препараты или кортикостероиды, могут потребоваться для облегчения симптомов или уменьшения повреждения тканей.
Например, как показано в Блоке 3, во время тяжелых системных инфекций с большим количеством присутствующих микроорганизмов высвобождаются высокие уровни патоген-ассоциированных молекулярных паттернов (PAMP), что приводит к чрезмерному производству цитокинов макрофагами, что может нанести вред организму.Кроме того, нейтрофилы начинают выделять свои протеазы и активные формы кислорода, которые убивают не только бактерии, но и окружающие ткани. Вредные эффекты включают высокую температуру, гипотензию, разрушение тканей, истощение, острый респираторный дистресс-синдром или ОРДС, диссеминированное внутрисосудистое свертывание или ДВС-синдром, повреждение эндотелия сосудов, гиповолемию и снижение перфузии крови через ткани и органы, приводящее к шоку, множественный системный орган. отказ (MOSF) и часто смерть.Этот чрезмерный воспалительный ответ называется синдромом системного воспалительного ответа, или ССВО, или каскадом шока.
Упражнение: Подумайте, пара, поделитесь вопросами
- Кратко опишите механизмы, которые позволяют замедлить кровоток в очаге инфекции и доставить фагоциты, белки комплемента и антитела в очаг инфекции.
- Почему так важно доставить плазму к месту заражения?
- Почему важно, чтобы диапедез происходил во время воспаления?
Хроническое воспаление также способствует развитию сердечно-сосудистых заболеваний, болезни Альцгеймера, диабета и рака.
- В случае рака предполагается, что, когда макрофаги продуцируют воспалительные цитокины, такие как TNF-альфа, эти цитокины активируют генный переключатель в раковой клетке, который включает синтез белков, которые способствуют репликации клеток и воспалению, одновременно блокируя апоптоз раковой клетки.